Workflows
 MDWeb is powered by a set of BioMoby Molecular Dynamics Web-Services (MDMoby).
MDWeb is powered by a set of BioMoby Molecular Dynamics Web-Services (MDMoby).
One of the main advantages of web services is the possibility to interconnect them building complex pipelines called workflows.
To facilitate most usual MD operations, MDWeb offers a collection of pre-packed workflows. Settings have been adapted to run successfully on most systems, and their use for non-experts is recommended. However, MDWeb provides also the same functionality as separate operations, so the individual parameters could be adjusted. The pre-packed operations include:
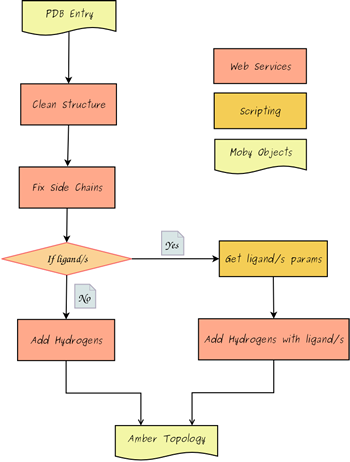
Workflows for generate topologies.
Workflows for solvate and neutralize structures.
Workflows for running a complete MD Setup.
Workflows for equilibrate systems.
This help section shows a short description of the workflows available. Clicking on the workflow name, a new window will automatically open, showing the workflow as a graphical diagram, where input object/s, output
object/s, web services and scripting pieces can be easily identified, together with the necessary interconnections.
Setup for Amber Simulation
|
|
Generate Topology for Amber.
- ForceField: Amber Parm99SB* for Proteins, Amber Parm99bsc0 for Nucleic Acids (DNA/RNA).
- Program: Leap from AmberTools Package.
- Remove crystallographic water molecules.
- Add hydrogen atoms and missing side chain atoms as appropiate.
|
|
|
Amber MD Setup. Structure Setup for AMBER Forcefield.
- ForceField: Amber Parm99SB* for Proteins, Amber Parm99bsc0 for Nucleic Acids (DNA/RNA).
- Programs: namd2 from NAMD Package, leap from AmberTools package, protpKa and CMIP.
- Generate Topology for AMBER.
- Protonate Histidine residues according to protpKa program algorithm.
- Add 20 water molecules at the energetically best favourable positions of the structure surface using CMIP program.
- Energy minimize hydrogen atoms for 500 steps of conjugate gradients, while the rest of the structure is kept fixed.
- Energy minimize the structure for 500 steps of conjugate gradients, restraining heavy atoms with a force constant of 50 Kcal/mol to their initial positions.
|
|
|
AMBER MD Setup with Solvation. Structure Setup + Solvation for AMBER Forcefield.
- ForceField: Amber Parm99SB* for Proteins, Amber Parm99bsc0 for Nucleic Acids (DNA/RNA).
- Programs: namd2 from NAMD Package, leap from AmberTools package, protpKa and CMIP.
- AMBER MD Setup.
- Set a truncated Octahedron box of TIP3P water molecules with a spacing distance of 15 Å around the system.
- Add Cl- and/or Na+ ions necessary to neutralize the system. Then, add the appropiate amount of ions up to a concentration of 50 mM.
- Further energy minimize the structure for 500 steps of conjugate gradients, restraining heavy atoms with a force constant of 50Kcal/mol to their initial positions.
|
|
|
AMBER Advanced Equilibration. System Equilibration.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Langevin Dynamics.
- Constant pressure dynamics via Nose-Hoover Langevin piston.
- SHAKE is used to maintain all bonds involving hydrogen atoms at their equilibrium values.
- Heat solvent to 300K. Solute atoms restrained (force constant of 10 Kcal/mol). Length 5ps.
- Reduce force constant to 5 Kcal/mol. Length 1ps.
- Reduce force constant to 2.5 Kcal/mol and limit restraints to backbone atoms. Length 1ps.
- Reduce force constant to 1 Kcal/mol. Length 1ps.
- Simulation without restraints. Length 1ps.
|
|
|
Complete Setup for AMBER forcefield (Structure Setup + Solvation + Equilibration).
- ForceField: Amber Parm99SB* for Proteins, Amber Parm99bsc0 for Nucleic Acids (DNA/RNA).
- Programs: namd2 from NAMD Package, leap from AmberTools package, protpKa and CMIP.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Langevin Dynamics.
- Constant pressure dynamics via Nose-Hoover Langevin piston.
- SHAKE is used to maintain all bonds involving hydrogen atoms at their equilibrium values.
- AMBER MD Setup with Solvation.
- AMBER Advanced Equilibration.
|
top
Setup for Gromacs Simulation
|
|
Generate Topology for GROMACS. Generate top and itp Topology Files for Gromacs.
- Programs: pdb2gmx from Gromacs Package.
- Remove crystallographic water molecules.
- Add side chain missing atoms using Leap from AmberTools package.
- Add Hydrogen atoms using pdb2gmx.
|
|
|
GROMACS MD Setup. Structure Setup for Gromacs.
- Programs: pdb2gmx, grompp, editconf, trjconv, make_ndx and mdrun from Gromacs Package, leap from AmberTools package, protpKa and CMIP.
- Generate Topology for GROMACS.
- Protonate Histidine residues according to protpKa program algorithm.
- Add 20 water molecules at the energetically best favourable positions of the structure surface using CMIP program.
- Energy minimize hydrogen atoms for 500 steps of hydrogen conjugate gradients, while the rest of the structure is kept fixed.
- Energy minimize the structure for 500 steps of structure conjugate gradients, restraining heavy atoms with a force constant of 500KJ/mol*nm2 to their initial positions.
|
|
|
GROMACS MD Setup with Solvation. Structure Setup + Solvation for Gromacs.
- Programs: pdb2gmx, grompp, editconf, trjconv, make_ndx, genbox, genion and mdrun from Gromacs Package, leap from AmberTools package, protpKa and CMIP.
- GROMACS MD Setup.
- Set a truncated Octahedron box of TIP3P water molecules (Amber FF) or SPC water molecules (other FF's) with a spacing distance of 15Å around the molecule.
- Add Cl- and/or Na+ ions necessary to neutralize the system. Then, add the appropiate amount of ions up to a concentration of 50 mM.
- Further energy minimize the structure for 500 steps of conjugate gradients, restraining heavy atoms with a force constant of 500KJ/mol*nm2 to their initial positions.
|
|
|
GROMACS Advanced Equilibration. System Equilibration.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Velocity-rescale algorithm.
- Constant pressure dynamics via Parrinello-Rahman algorithm.
- LINCS Linear Constraint Solver is used to maintain all bonds at their equilibrium values.
- Heat solvent to 300K. Solute atoms restrained (Force constant of 400 KJ/mol*nm2. Length 5ps.
- Reduce force constant to 300 KJ/mol*nm2. Length 1ps.
- Reduce force constant to 200 KJ/mol*nm2 and limit restraints to backbone atoms. Length 1ps.
- Reduce force constant to 100 KJ/mol*nm2. Length 1ps.
- Simulation without restraints. Length 1ps.
|
|
|
GROMACS FULL MD Setup. Complete Setup for Gromacs Package (Structure Setup + Solvation + Equilibration).
- Programs: pdb2gmx, grompp, editconf, trjconv, make_ndx, genbox, genion and mdrun from Gromacs Package, leap from AmberTools package, protpKa and CMIP.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Velocity-rescale algorithm.
- Constant pressure dynamics via Parrinello-Rahman algorithm.
- LINCS Linear Constraint Solver was used to maintain all bonds at their equilibrium values.
- GROMACS MD Setup with Solvation.
- GROMACS Advanced Equilibration.
|
top
Setup for NAMD Simulation
|
|
Generate Topology for NAMD. Generate PSF Topology for Charmm Forcefield.
- ForceField: Charmm-27.
- Program: psfgen from NAMD Package.
- Warning: Ligands not allowed.
- Remove crystallographic water molecules.
- Add hydrogen atoms and missing side chain atoms as appropiate.
|
|
|
NAMD MD Setup. Structure Setup for Charmm Forcefield.
- ForceField: Charmm-27.
- Programs: psfgen, vmd (solvate and autoionize plugins) and namd2 from NAMD Package, protpKa and CMIP.
- Generate Topology for NAMD.
- Protonate Histidine residues according to protpKa program algorithm.
- Add 20 water molecules at the energetically best favourable positions of the structure surface using CMIP program.
- Energy minimize hydrogen atoms for 500 steps of conjugate gradients, while the rest of the structure is kept fixed.
- Energy minimize the structure for 500 steps of conjugate gradients, restraining heavy atoms with a force constant of 50Kcal/mol to their initial positions.
|
|
|
NAMD MD Setup with Solvation. Structure Setup + Solvation for Charmm Forcefield.
- ForceField: Charmm-27.
- Programs: psfgen, vmd (solvate and autoionize plugins) and namd2 from NAMD Package, protpKa and CMIP.
- NAMD MD Setup.
- Set a cubic box of TIP3P water molecules with a spacing distance of 15 Å.
- Add Cl- and/or Na+ ions necessary to neutralize the system. Then, add the appropiate amount of ions up to a concentration of 50 mM.
- Further energy minimize the structure for 500 steps of conjugate gradients, restraining heavy atoms with a force constant of 50Kcal/mol to their initial positions.
|
|
|
NAMD Advanced Equilibration. System Equilibration.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Langevin Dynamics.
- Constant pressure dynamics via Nose-Hoover Langevin piston.
- SHAKE is used to maintain all bonds involving hydrogen atoms at their equilibrium values.
- Heat solvent to 300K. Solute atoms restrained (force constant of 10 Kcal/mol). Length 5ps.
- Reduce force constant to 5 Kcal/mol. Length 1ps.
- Reduce force constant to 2.5 Kcal/mol and limit restraints to backbone atoms. Length 1ps.
- Reduce force constant to 1 Kcal/mol. Length 1ps.
- Simulation without restraints. Length 1ps.
|
|
|
NAMD FULL MD Setup. Complete Setup for Charmm Forcefield (Structure Setup + Solvation + Equilibration).
- ForceField: Charmm-27.
- Programs: psfgen, vmd (solvate and autoionize plugins) and namd2 from NAMD Package, protpKa and CMIP.
- Equilibration steps done in NPT ensemble with Periodic Boundary Conditions.
- Particle Mesh Ewald (PME) for full-system periodic electrostatics.
- Constant temperature dynamics via Langevin Dynamics.
- Constant pressure dynamics via Nose-Hoover Langevin piston.
- SHAKE is used to maintain all bonds involving hydrogen atoms at their equilibrium values.
- NAMD MD Setup with Solvation.
- NAMD Advanced Equilibration.
|