Ligand Checking
The majority of the structures in the Protein Data Bank have a non-standard residues (known as heteroatoms or ligands). Molecular dynamics force-fields are designed to work with standard amino acids or nucleotides. To include a ligand in the simulation, a complete description of the molecule as a parameterization file must be provided.
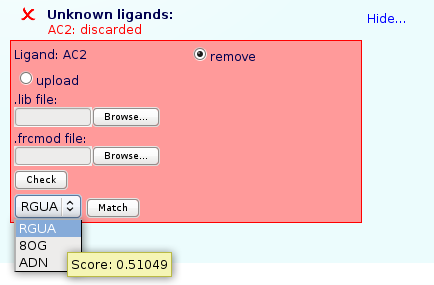
Ligands are divided in two groups: Known and Unknown, depending on the ligand parameters availability.
Known ligands correspond to a database of heteroatom used on our MoDEL library, and can be incorporated to the simulated system. 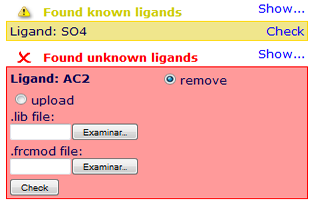 For the unknown ones, NAFlex gives the user the possibility to upload parameter files (in Amber format, lib/frcmod).
For the unknown ones, NAFlex gives the user the possibility to upload parameter files (in Amber format, lib/frcmod).
If parameter files are not uploaded, the ligand will be removed from the structure. Please note that to avoid inconsistent systems, the default option is to remove ligands, even the known ones.
For Unknown Ligands, a tanimoto coefficient against all our MoDEL library is run, giving the user the three more similar molecules found in our database. To know the Tanimoto coefficient user just have to hold the mouse pointer over the ligand name for a second.
Please note that choosing a similar ligand could raise to setup problems depending on the differences between the ligand in the input structure and the similar one.
Atom name match window
Finally, in both groups (known or unknown), atom naming of the ligand needs to be checked.
Atom names in non-standard residues found in the PDB structure can differ to those in the available parameter files. To generate a consistent structure, atom names should match.
To make this process easier, NAFlex opens a new window with two synchronized JMol applets, showing the incoming (PDB) ligand structure (left window) and the library ligand (right window).

If the two ligands are siimilar to each other, both structures will be showed with the same orientation, and rotation of the structures will be synchronized.
In the middle of JMol windows, there is a matching table. The first column shows the ligand atom names as appear in the incoming PDB file, while the second one shows the atom names contained in the ligand parameter file.
For atoms having different names, user can use this central form to do the necessary corrections. Atoms located under cursor are highlighted in the corresponding JMol applet, thus allowing the correct identification.
 This process can only be skipped when the ligand is mono-atomic, as in the case of ions (Auto link).
This process can only be skipped when the ligand is mono-atomic, as in the case of ions (Auto link).