----
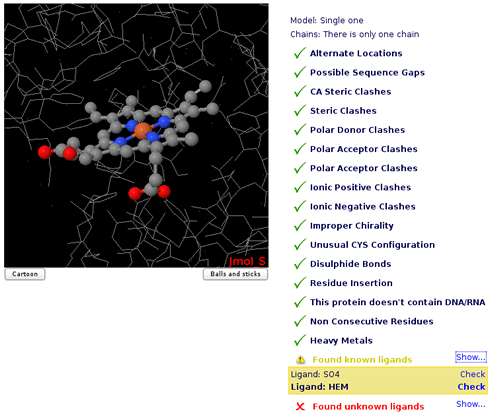
Structure Checking
In a Molecular Dynamics simulation, the correctness of input structures is crucial. Small errors in the input structure may cause MD simulations to became unstable or give unrealistic trajectories.

The purpose of the initial Structure checking page is to check for the most common problems in the input of MD simulations, allowing the user to select possible solutions when available. Besides, structure checking allows to select fragments of the system to be simulated in the case that alternate models or multiple subunits are present in the incoming structure.
An interactive JMol applet window provides additional help in assessing the significance of errors found. Please note that MD itself can correct some of the problems found (specially steric clashes), but other can be only be corrected by editing the structure beforehand.
The checking consists in a list of possible options to choose for the system to be simulated:
- Structure Model.
- Structure Chain/s.
- Residue/Atom Alternate Locations.
- Sequence Gaps/Non Consecutive Residues.
- Atom Clashes: Steric, Alpha-Carbon, Polar Donor, Polar Acceptor, Apolar, and Ionic Positive/Negative.
- Thr Improper Chirality.
- Unusual peptide-bond cis configuration.
- Disulphide Bonds.
- Residue Insertions.
- Structure containing DNA/RNA.
- Metals.
- Ligands.
 Ok: no problems found.
Ok: no problems found. Warning: important information or options to choose.
Warning: important information or options to choose. Error: will probably cause MD failure.
Error: will probably cause MD failure.
Choosing Structure Model
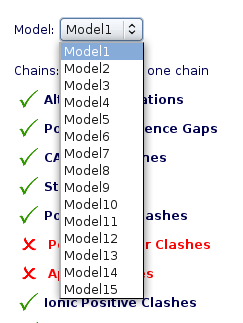
For PDB files containing multiple models (common for instance in NMR obtained structures), only one of the models can be simulated. The interface shows you the list of possible structure models to choose.
When chosen, all checking parameters are automatically recomputed with the selected model..
Choosing Structure Chain/s
For structures containing several Chains, user can select the ones to be included in the simulation. A Jmol applet helps in the identification of the chains, highlighting them when passing over the name with the mouse pointer. Be careful that selecting non complete structures may lead to unrealistic MD trajectories.

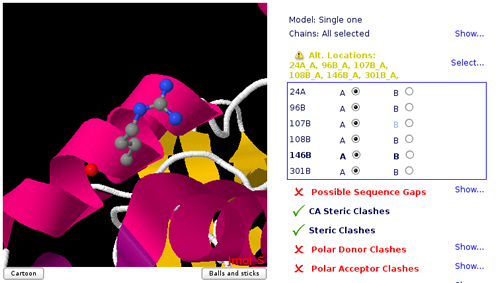
Choosing Atom/Residue Alternate Locations
Alternate Location indicators are used for atoms where more than one position is detected.
Within a residue, all atoms that are associated with each other in a given conformation are assigned the same alternate position indicator.
User can choose the residue alternate location of interest helped by a Jmol visualizer applet.
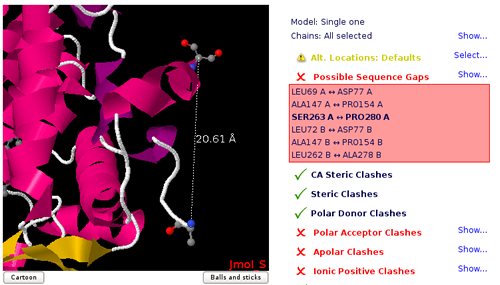
Sequence Gaps/Non Consecutive Residues
Flexible protein regions can have too low electron density to be detected in X-Ray diffraction experiments, and can be missing from PDB structures. This situation gives unrealistic structures where protein is split in several unconnected chains with new N- and C- terminal residues and buried protein regions become exposed to solvent. Simulation of such structures will give untrusty results and should be corrected by filling the missing gaps. MDWeb detects gaps both from the residue numbering and the existence or non-realistic bond distances.
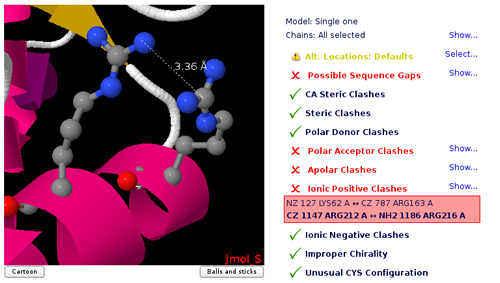
Atom Clashes
Atoms that are too close in space can have a problem of energetic repulsion. MDweb provides the list of atom pairs and the corresponding distances that can have potential problems. Most of clashes come from over-compactation of crystal structures and are naturally corrected on system setup or MD equilibration, but may lead to a significant distortion of the structure. Clashes are classified in different groups, depending on the atom types involved:
| Group | Description | Distance cut-off |
| Steric Clashes | Any atom pair | 1 Å |
| CA Steric Clashes | Alpha-carbon atom pairs | 3.8 ± 1Å |
| Polar Donor/Acceptor Clashes | Polar hydrogen bond donor/acceptor atom pairs. | 3.1 Å |
| Note that common polar clashes come from mis-assignment of side chain atoms in Asn, Gln, or Thr residues. | ||
| Apolar Clashes | Apolar atoms | 2.9 Å |
| Note that possible apolar clashes can come from atoms neighbouring legitimate hydrogen bonds. | ||
| Ionic Positive/Negative Clashes | Positively/negatively charged atoms | 3.5 Å |
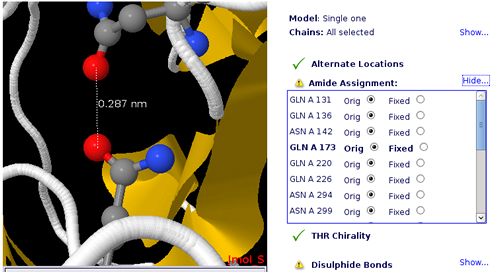
Improper Amide Assignment
Amide Groups in the side chains of Glutamine and Asparagine (Gln, Asn) residues can act simultaneously as hydrogen bond donors and acceptors. The electron density near the nitrogen and oxygen atoms of these functional groups is compatible with two rotamers related by a 2-fold symmetry axis. Therefore, electron density maps obtained from X-ray diffraction experiments can be wrongly interpreted leading to improper amide assignments.
User can choose the best fitted amide group configuration helped by Jmol applet.
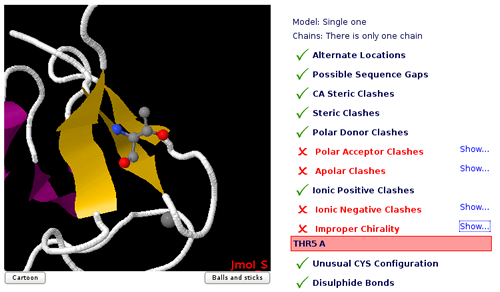
Improper Chirality
Chiral molecules lack an internal plane of symmetry and thus cannot be superimposed on
their mirror images.
Organic molecules which contain at least one tetrahedral carbon atom bound to four different substituent groups are chiral. That carbon atom is a "chiral centre".
Alpha carbons of all amino acids except glycine are chiral, but in natural proteins only one of the configuration is found
(L-configuration). D-amino acids are not naturally found in proteins.
Threonine and Isoleucine side chains contain an additional chiral centre at the beta carbon. Mis-assignment of atom names in the structure may lead to an improper chirality.
Usual restrictions in molecular dynamics force-fields do not allow to reverse chiral centers. MDWeb checking page will help you identifying these improper chiralities.
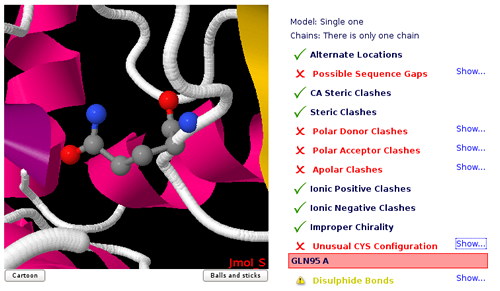
Unusual cis/trans Configuration
Cis/trans isomerism describes the orientation of functional groups within a molecule where a bond has a limited possibility of rotation.
Peptide bonds have a considerably double bond character and present cis/trans isomerism. Nearly all peptide bonds appear in the trans, whereas cis configuration is sometimes found in Proline residues. Omega torsion angle (ω) is computed to identify unusual cis/trans configurations in peptide bonds.
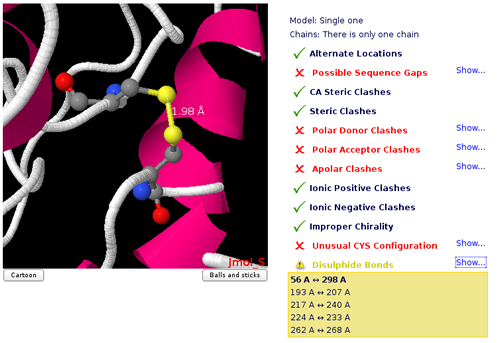
Disulphide Bonds
Disulphide bonds are covalent bonds usually formed by a pair of thiol groups. They are also called SS-bonds or disulphide bridges.
As they are covalent bonds, it is of great importance in a simulation to take them into account.
In fact, SS-bonds are known to have an important structural role in protein structure and stability.
Setup procedures can detect and link thiol groups forming disulphide bridges, but it is important to know whether the structure contains such covalent bonds.
Jmol applet will show the structure's disulphide bonds, as well as the distance between the sulphur atoms involved.
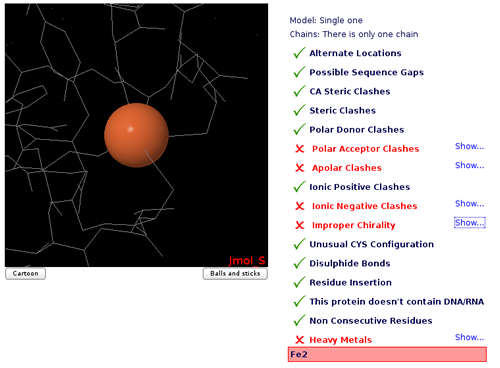
Metals
Metals (Mg, Zn, Mn, Mo, Ni, Fe, Co, Cu, Hg, Cd, Ag, Au) are usually found making coordination complexes with protein residues.
A typical example is the so called Zinc finger, where a Zn ion is coordinated by cysteines and histidine residues, forming a structural motifs that help stabilize the structure. Metall complexes involve a complex chemistry and are not normallly covered by standard force-fields as they require a complete re-parametrization of both metal and ligands. Simulations of such complexes would normally require the setting of distance restrains to maintain the coordination structure.
Structure containing DNA/RNA
MDWeb can work not only with proteins but also with nucleic acids and even with protein-DNA and protein-RNA complexes, however this feature has not been extensively tested.
The structure checking process informs about the existence of nucleic acids in the input structure. Removal of nucleic acids if desired can be done in the chain selector.

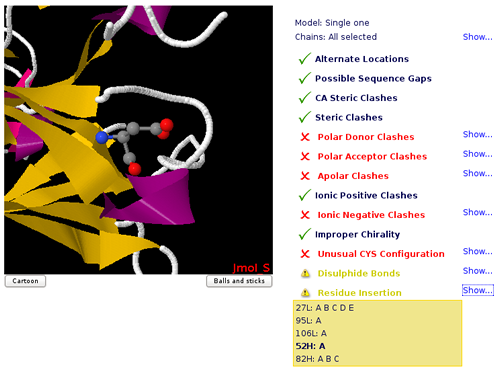
Residue Insertion
PDB specification contains an optional field, named Residue insertion code to allow structure providers to match residue numbering between proteins from different sources when insertions and deletions occur. Simulation setup will remove such field and renumber residues accordingly.
The MDWeb checking page shows the set of residues with insertion codes in the Jmol applet.
Ligands
The majority of the structures in the Protein Data Bank have a non-standard residues included, identified as "heteroatoms" in the structure. Molecular dynamics force-fields contain parameters for standard amino acids and nucleotides but not normally for such compounds. To include a ligand in the simulation, the complete description of the ligand structure and the corresponding force-field parameters should be provided. MDWeb contains a extense library of already parameterized ligands that can be included in the simulation ("known ligands"). In the checking step, the presence of ligands in the structure, as well as the availability of the corresponding parameters library will be tested. In the case of unknown ligands, not available in the library, used will be prompted for the corresponding parameter data.
Please note that the default behaviour of MDWeb checking phase is to remove ligands from the structure.