


BIGNASim database structure and analysis portal for nucleic acids simulation data
----
DNA & RNA Structure and Helical Parameters Analyses (NAFlex)
----Tutorial 2 -- Global analysis (XCGY)
Tutorial 3 -- Meta-trajectory (XCGY)
Tutorial 4 -- Experimental vs MD analysis
----Tutorial 1: Search (DDD)
This tutorial shows several real examples on how to look for a particular nucleic acid sequence, nucleic acid fragment or base pair step in the BIGNASim database using its search engine. The search section of the portal is accessed through the main menu:

The search section contains three different possibilities (see this help searching section for more details):
- Search by sequence or specific sequence fragments (using regular expressions)
- Search by specific base-pair-steps (with or without flanking regions)
- Search by an extensive nucleic acid ontology (as defined here)
Now let's supose that we are interested in finding three different types of information from the database:
- Information related to the well-known Drew-Dickerson Dodecamer (DDD)
- Information related to the AT Base-Pair Step (central in DDD)
- Information related to a naked duplex B-DNA structure with a particular nucleotidic fragment
Drew Dickerson Dodecamer (DDD)
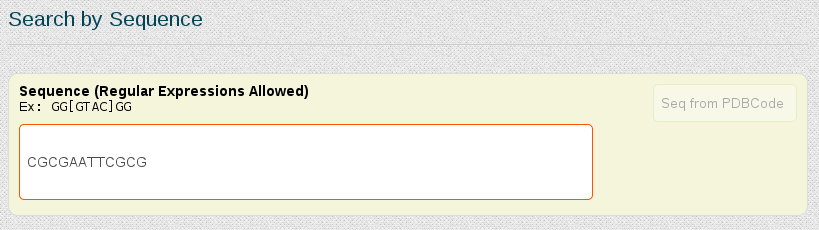
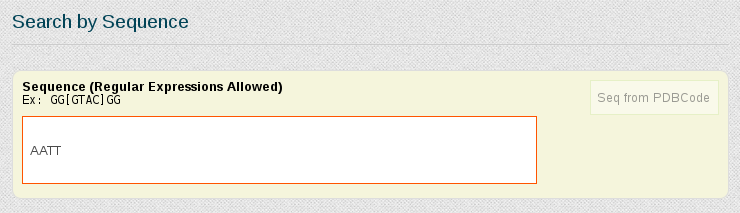
In this case, as we already know the nucleotidic sequence of the DDD (CGCGAATTCGCG), we can just search for the complete sequence in the "Search by Sequence" section of the portal:
We have another possibility here, as it can be considered a special sequence because of its importance, it is included in our Sequence Features section of the nucleic acids ontology:

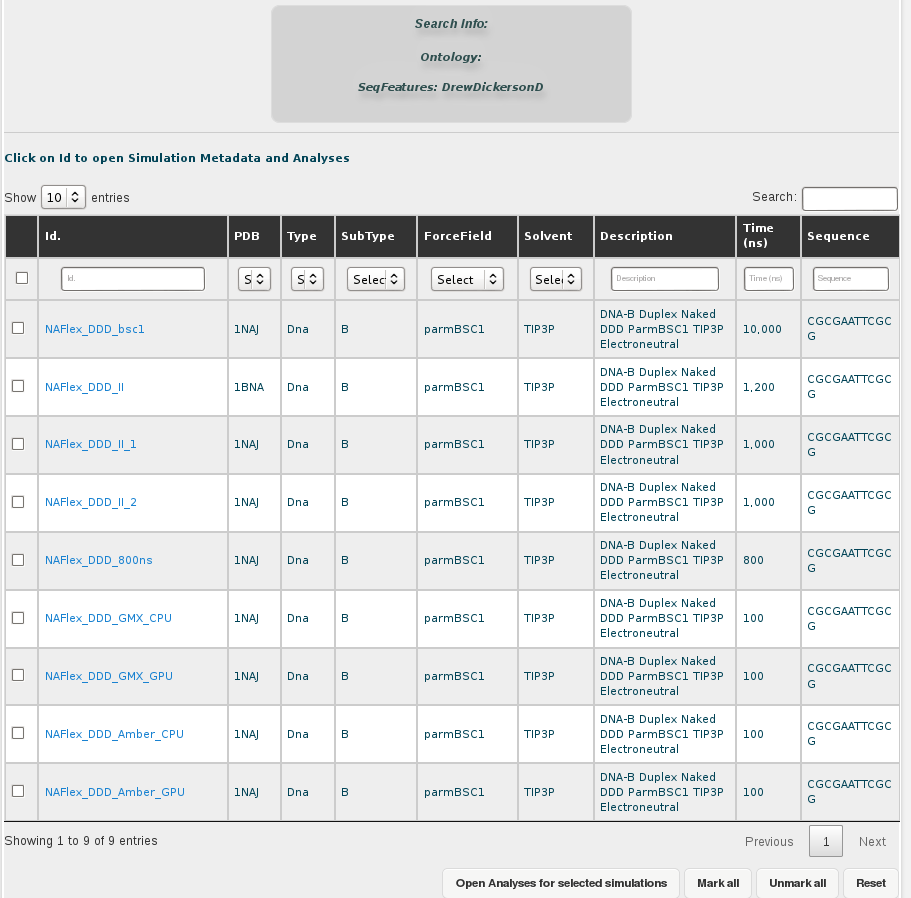
Both possibilities open a browse page with all the simulations stored in our database for this particular sequence. In this case, we have 5 different trajectories, the longest one having 10µseconds. Now we could open each of the simulations individually to look at the MD simulation metadata and trajectory analyses or obtain global information from more than one simulations together, selecting the desired entries and clicking at Open analyses for selected simulations (see browsing section for more information).
AT Base-Pair Step (central in DDD)
If we are interested in the DDD central base-pair step CGCGAATTCGCG, the Search by Base Pair Step section of the search engine can be used:
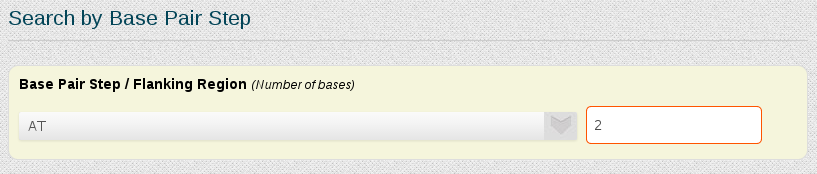
In the selector we just have to choose the desired base-pair step, in this case AT. There's also the possibility to add a number of wanted flanking nucleotides, to ensure that information obtained will not be from base-pair steps placed at terminal regions, typically showing distorted flexibility parameters. In this example we will put two nucleotides of flanking region. The same procedure can be applied for every base-pair step.
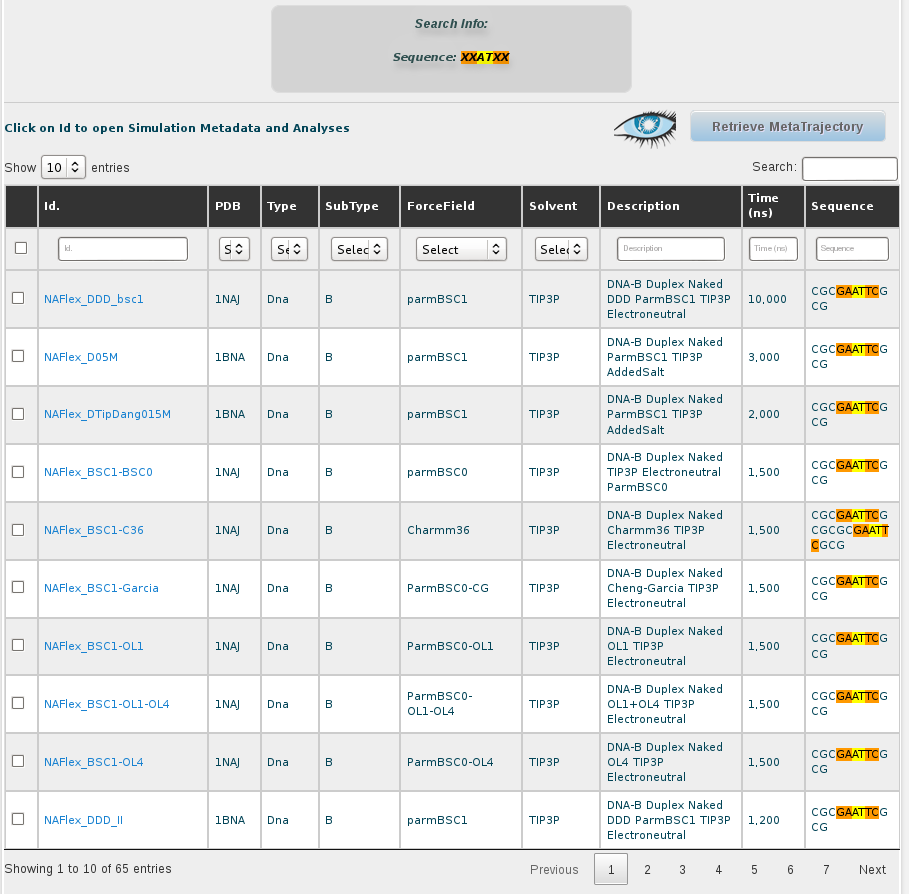
The results obtained for the AT base-pair step with the current content of the database are 51 different simulations. Looking at the sequence column, the interesting AT pair, together with the flanking region, can be easily identified thanks to the marking in yellow and orange color, respectively. The first thing we can see in this browse page is that there are different sequences stored in the database other than the DDD containing the AT base pair, specifically 46 sequences (at the moment of writing this tutorial), some of them having more than one occurrence of it. That offers enough information to compare between the flexibility parameters obtained for just the 5 sequences of DDD obtained in the previous section of this tutorial with the rest of sequences having the AT base pair. To obtain flexibility information for just the sequences other than DDD, we can click at the selector of records shown in the browse page, at the top-left part of the screen, and choose 100 records:
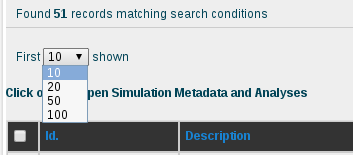
Once we have all the records on the screen, we can select them all by clicking at the checkbox placed at the left part of the table header, next to the Id. title, and then uncheck the ones corresponding to the DDD sequence. The selector of records placed in the top part of the Id. column will help in finding the 5 sequences having DDD in their name:
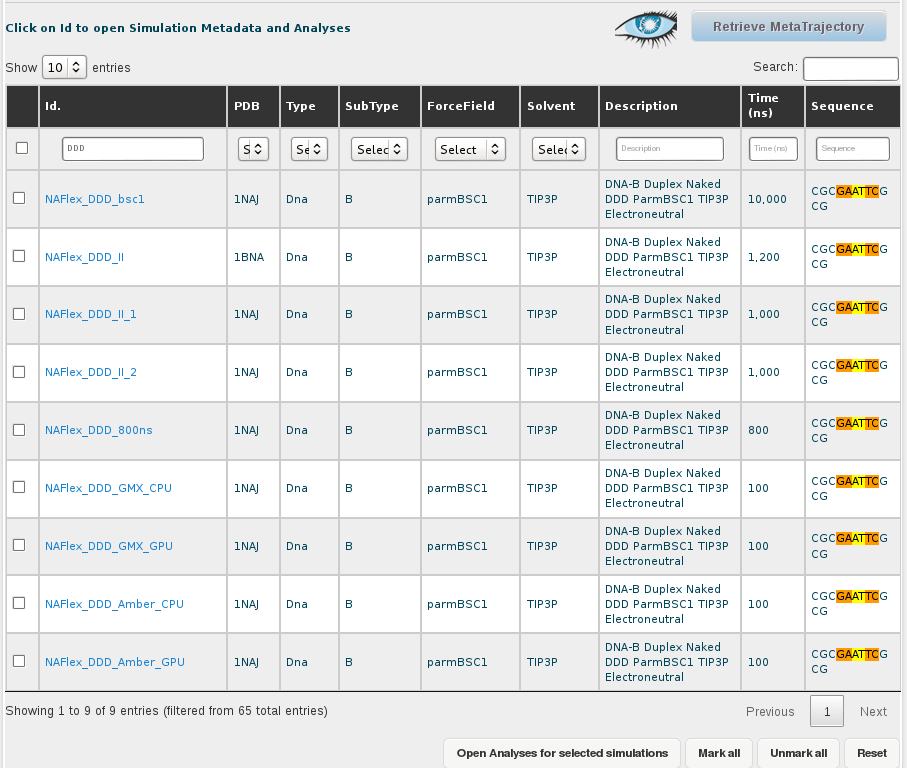
The final step consists on clicking at the Open Analyses for selected simulations button at the bottom of the browse page, which will send us to the analysis section of BIGNASim (see analysis section).

Now in this section, we can click at the AT base-pair step button, and then open every analysis we are interested in. In order to compare the results with the ones corresponding to the AT base-pairs from just DDD sequences, we can repeat the previous steps (or repeat the steps done in the first section of this tutorial) to retreive the analysis for these particular sequences. The idea is to get to the same analyses page, click at the AT base-pair button, and then compare the results with two different browser windows.
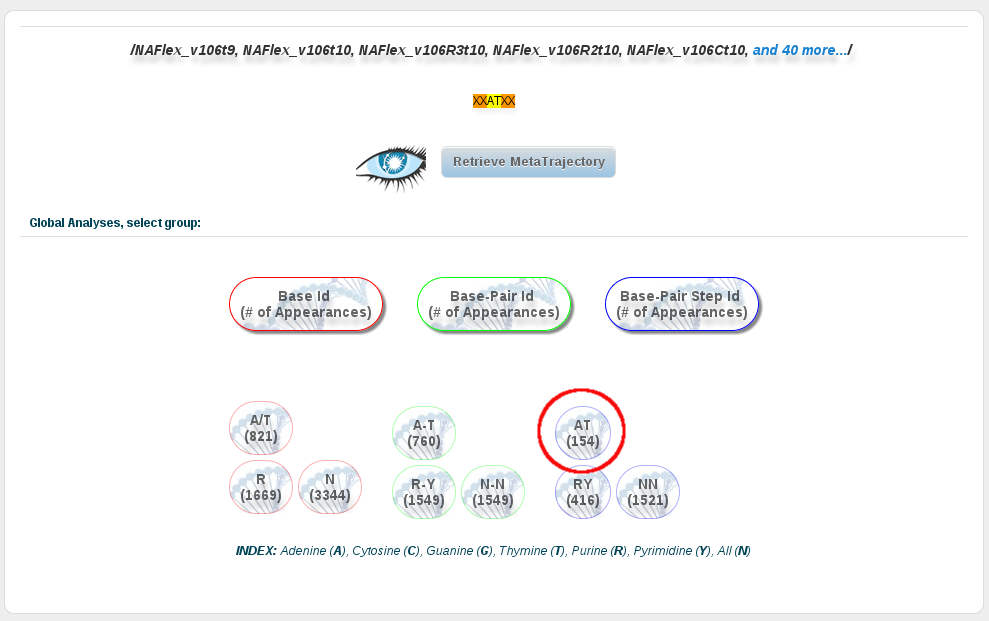
To obtain more information about the analyses included in the analyses page, and how to use it, please refer to the analysis section or the analysis tutorial of this help.
Naked duplex B-DNA structure with a particular nucleotidic fragment
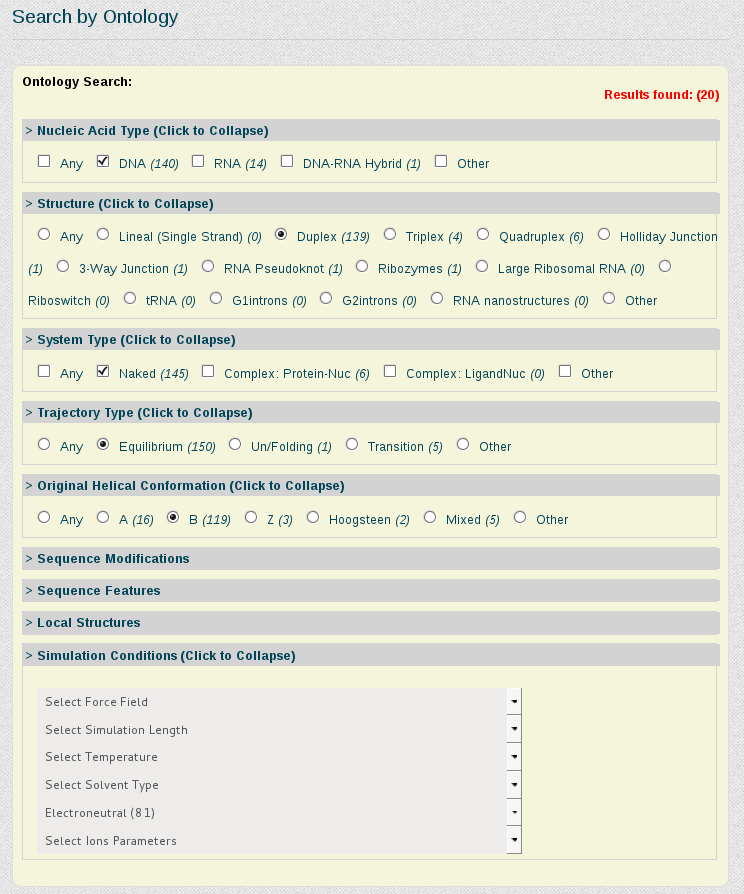
Now suppose we want to be more specific about the simulations we are interested in study, and we just want to retrieve those trajectories having the DDD central tetramer (AATT) computed on naked B-DNA duplex structures, simulated in equilibrium conditions and electroneutral charge. For that, we need to write the AATT tetramer in the Search by Sequence section, and refine the search using the Search by Ontology section.
In the Search by Ontology section, we can go branch by branch refining the search. In this case, we will click DNA in Nucleic Acid Type area, Duplex in Structure area, Naked in System Type area, Equilibrium in Trajectory Type, B in Helical Conformation and finally Electroneutral in Simulation Conditions, Ionic Concentration. Every time a search parameter is chosen, the search engine computes the number of results stored for the current selected refinement specification and shows it on-the-fly in the top right part of the Search by Ontology section, in red color.
Again, the results are shown in a browse page. In the description we can see that results are indeed duplex naked B-DNA structure simulations, as defined in the search. Still, results obtained contain a sequence different than the DDD having the tetramer we were interested in (AATT): 1rvh (GCAAAATTTTGC). For the rest of DDD trajectories, the differences rely on the particular simulation parameters used in the MD, e.g. solvent type, ionic parameters or total length.
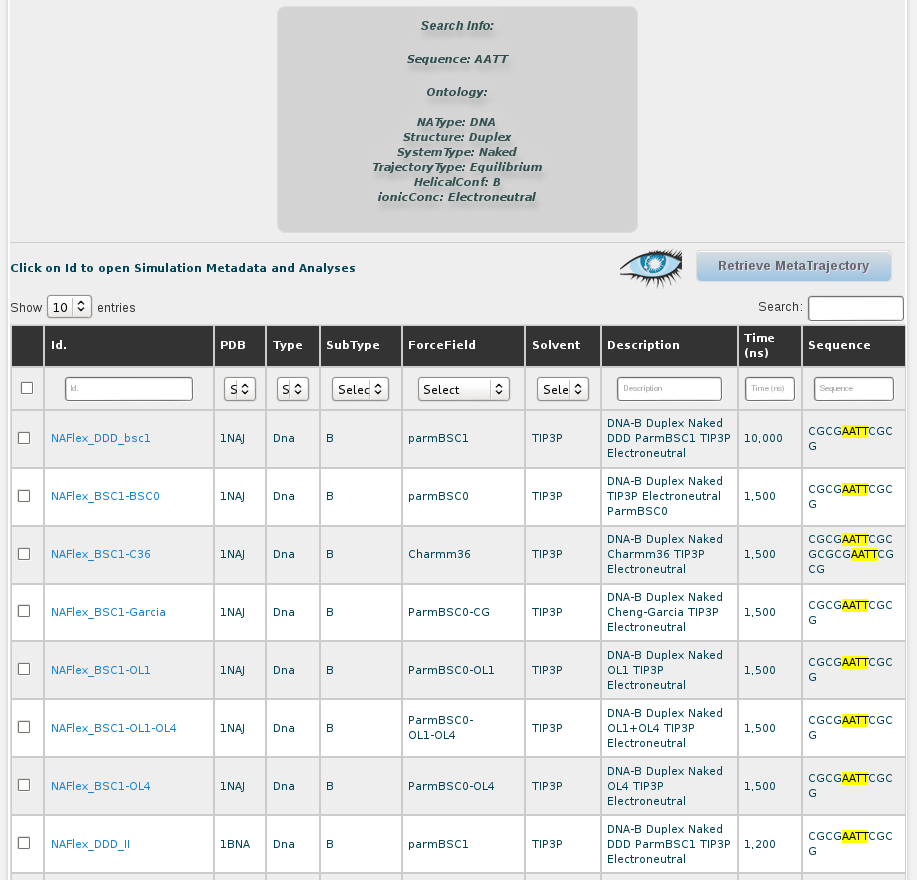
Now, with this results, we can go one by one to inspect flexibility properties for each of the simulations independently (see simulation section of this help), we can analyse the global flexibility of our AATT tetramer selecting all the trajectories and clicking at Open Analysis for selected simulations button, or we can generate a meta-trajectory with this particular nucleotidic fragment joining together atomic coordinates of a selection set of simulations from all the resulting ones shown in the browse page. More details on how to build a meta-trajectory with BIGNASim can be found at the meta-trajectory tutorial and the meta-trajectory section in this help.