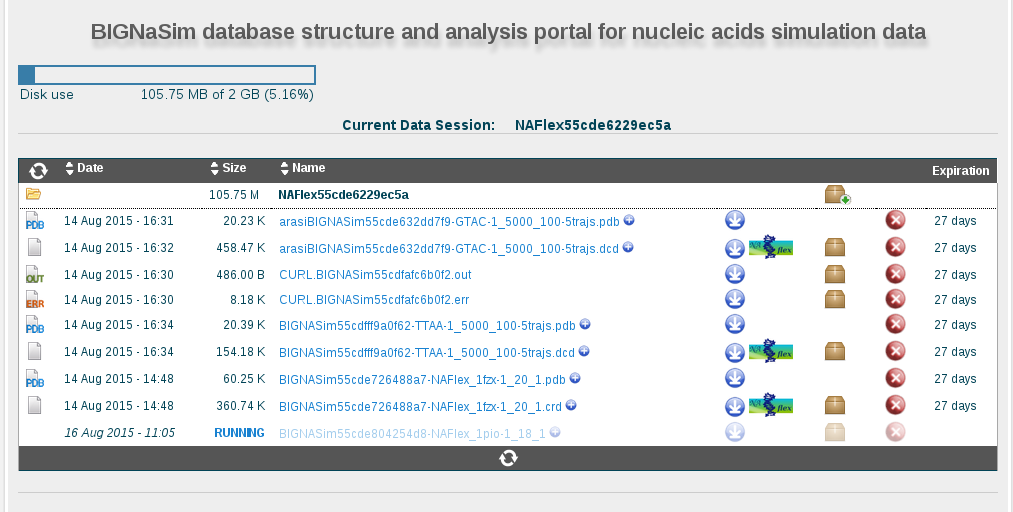
BIGNASim database structure and analysis portal for nucleic acids simulation data
----
DNA & RNA Structure and Helical Parameters Analyses (NAFlex)
----Tutorial 2 -- Global analysis (XCGY)
Tutorial 3 -- Meta-trajectory (XCGY)
Tutorial 4 -- Experimental vs MD analysis
----Tutorial 3: Meta-trajectories (XCGY)
This tutorial shows how to build a meta-trajectory containing a particular nucleotidic fragment with BIGNASim portal. For this tutorial, we will continue with the real example presented in the previous global analyses tutorial: CG base-pair step and the influence of the tetranucleotide environment in the sequence-dependent polymorphism of particular base-pair steps (P. Dans et al, NAR 2014). We have seen in the previous tutorial how to extract directly from the database the population of a particular helical parameter, recovering the Twist bimodality found in the above mentioned research. However, one can be interested in study different properties, not stored in BIGNASim database. For that, there is a possibility to generate and download (and also send to NAFlex server) a meta-trajectory containing just the nucleotide fragment of interest. It is build joining together the atomic information (3D cartesian coordinates) from all the simulations stored in our database enclosing the fragment. To illustrate the power of BIGNASim database, we interrogated the DB searching for the 16 possible different tetramers including CG base-pair, and we obtained results for all of them, going from 2 (TCGG) to 49 (TCGC) occurrences. We must say that for now, BIGNASim is populated with just a set of trajectories generated during the development and validation of the parmBSC1 force-field, and the information stored is going to be increased with new simulations.
In this tutorial, we will generate a meta-trajectory of one of the tetramers having less occurrences (ACGT, 4 occurrences), in order to show a quick example of the steps to be followed. The same example could be used to generate meta-trajectories for all of the possible tetranucleotides including CG, thus obtaining a valuable set of ensembles to analyse and compare. The first step in this tutorial consists on accessing the search section of the portal from the main menu:

The search section contains three different possibilities (see this help searching section for more details):
- Search by sequence or specific sequence fragments (using regular expressions)
- Search by specific base-pair-steps (with or without flanking regions)
- Search by an extensive nucleic acid ontology (as defined here)
To generate a ACGT meta-trajectory, we will use the Search by Sequence Fragment section of the search engine. The Search by Base-Pair Step field can also be used if the particular sequence fragment we are interested in is precisely a base-pair step.
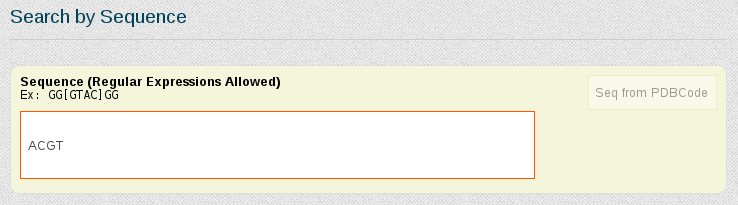
As we are interested in studying the behavior of the tetramer in just normal duplex B-DNA structures, simulated in equilibrium conditions, we select the corresponding features from the Search by Ontology section:
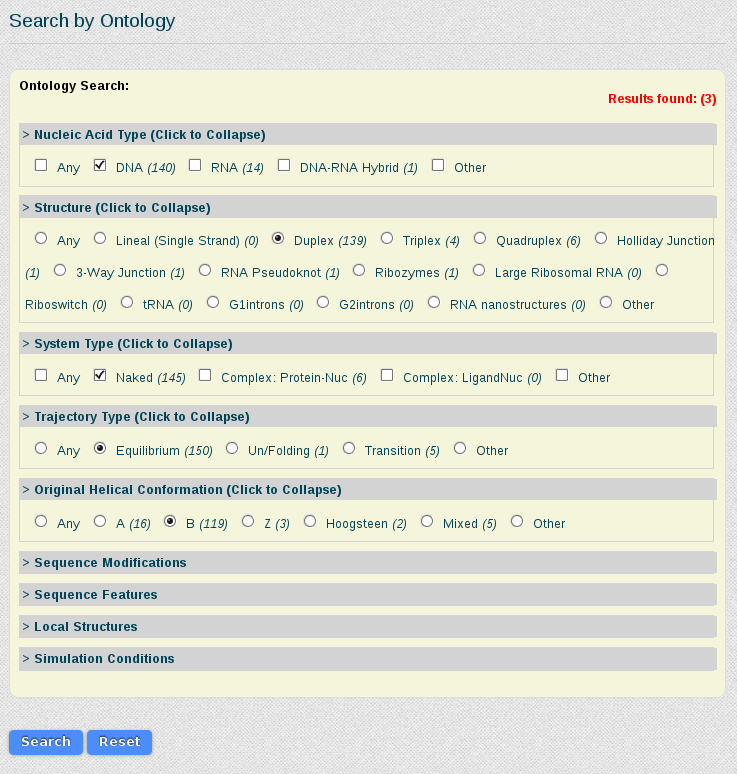
Results for this search are 3 different simulations (one less because of the filtering done). The information that we will have in the final meta-trajectory is going to be the sum of all these trajectories, so it will have information equivalent to 1.290µs (500 + 500 + 290 ns). Note that if we would have taken the most represented tetramer including CG (TCGC), we would be generating a meta-trajectory corresponding to up to 39 µs of simulated time. BIGNASim offers the possibility to generate a meta-trajectory combining all these different simulation trajectories. It is built getting the atomic information (3D-coordinates and topology) of the selected nucleotidic fragment. In this case, a meta-trajectory for the ACGT tetramer, representing 1,290µs of simulation data will be generated.
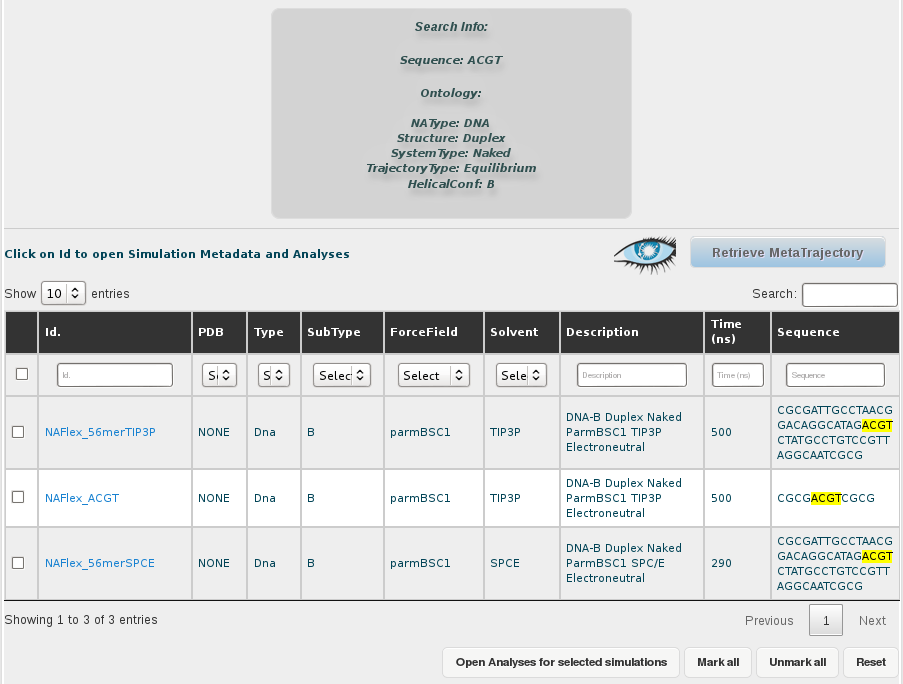
For that, the first thing we need is to select all the trajectories obtained in the search. That implies two different steps, first show all the results in the browse page, selecting an appropiate number of rows to be displayed in the selector placed in the top-left corner of the screen. In this case, as we are just dealing with 3 different trajectories, that is not necessary, but we should keep that in mind when working with a higher number of results.
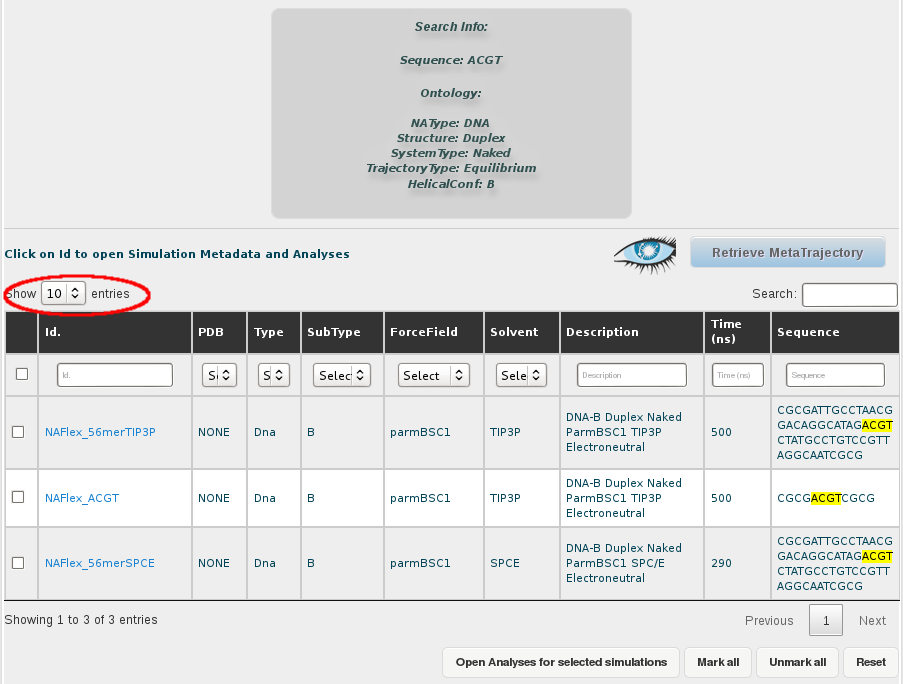
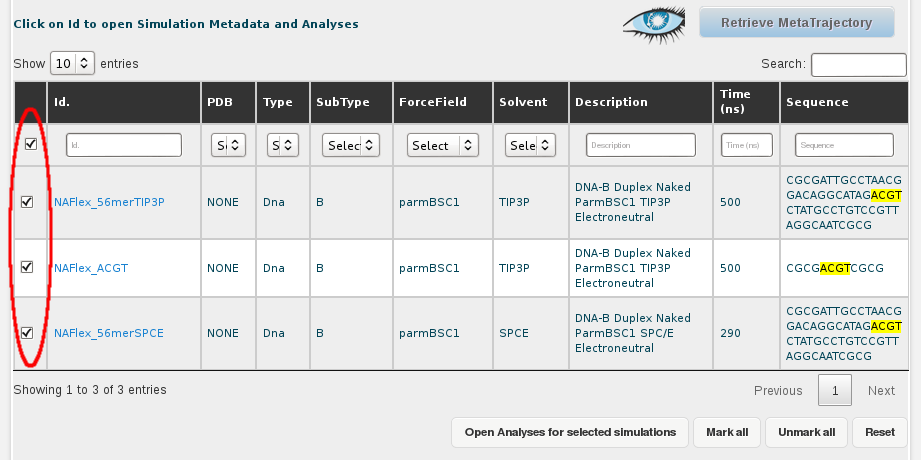
Then, we select all the simulations (or just the ones we are interested in), clicking at the respective checkboxes. A single click to the checkbox placed in the top-left corner of the table, next to the Id. title will automatically select all trajectories. This can also be done with a button placed at the bottom of the browse page named Mark All.
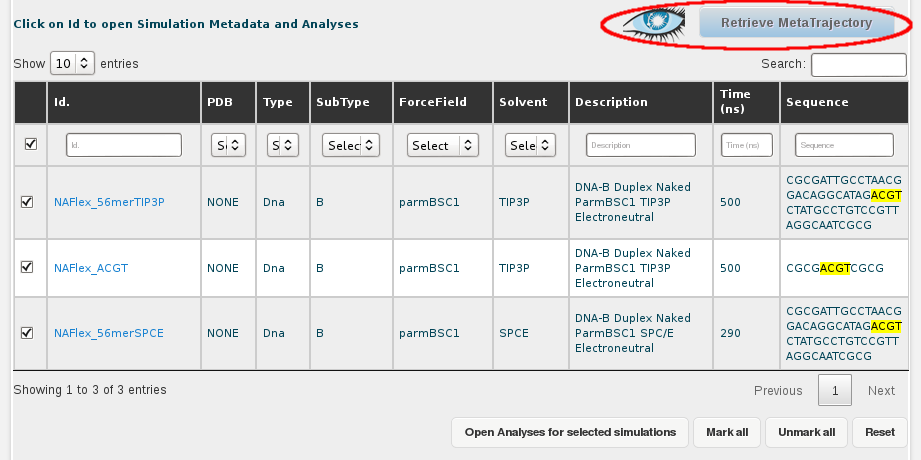
Once we have the desired trajectories selected, the next step is clicking at the Retrieve Metatrajectory button placed at the top-right part of the screen (next to the Cassandra's logo, representing an eye).
The next step for generating the meta-trajectory is to define a desired frame step. In this case, we want to extract 100 snapshots as representatives of each trajectory. For that, we type in the Frame input section: 1:5000:50 (for snapshot 1 to snapshot 5000, every 50 snapshots). At this step, we can choose between downloading the new generated trajectory or send it directly to the NAFlex nucleic acid flexibility server. As this trajectory is going to be quite big, we will download it to work in our own machines. The download section offers different possible trajectory formats. We will choose the one by default, DCD binary, because is one of the most lighweight formats, and it will accelerate the process.

The downloading process will be launched asynchronously. A waiting page will be opened informing that the trajectory is currently being generated. This waiting page will be automatically redirected to a data manager workspace where the resulting trajectory (as well as all the trajectories generated in this browser session) can be found, downloaded, or sent to NAFlex server (see data manager help section).