Bioactive Conformational Ensemble Help - Introduction
Introduction
BCE Strategy
Exploring the conformational preferences of small flexible ligands plays an increasingly important role in drug design. Accurate and efficient bioactive conformational search is one of the most fundamental topics in computer-aided drug design. Because bioactivity is based on binding, and binding is based on spatial complementarity, the prediction of the three-dimensional (3D) shape of a molecule is essential. Estimating the relative free energy of a ligand in its target- bound state (i.e. the bioactive conformation) is necessary to understand the process of molecular recognition, to optimize the potency of bioactive molecules and to improve the accuracy of structure-based drug design methods.
Since the bioactive conformation of the ligand may differ from the global minimum of the free ligand in the physiological environment, one has to evaluate the energetic cost required for adopting the bioactive conformation. 1,2,3,4
Previous studies indicated that multilevel strategy 5 represent an efficient methodology for the conformational search of drug-like We combined low-level (LL) method for sampling the conformational minima and high-level (HL) ab-initio calculations for estimating their relative stability. The multi-level strategy was automated and tested on various ligands with different numbers of atoms, charge and rotatable bonds.
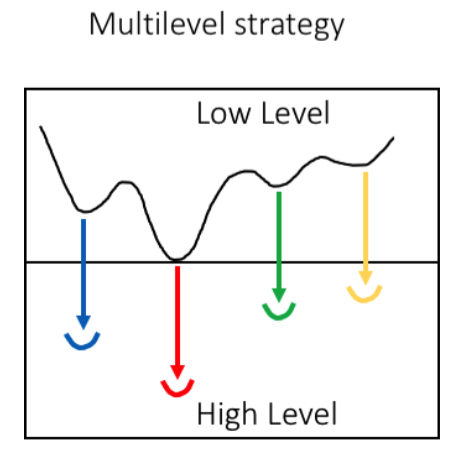
The proposed strategy represents an efficient tool for predicting the conformational landscape of drugs while keeping a reasonable balance between chemical accuracy and computational cost. Bioactive Conformational Ensemble (BCE) is a web-based tool for the for analysis of bioactive drug conformations and the flexibility of ligands in solution. The server incorporates powerful protocols for prediction and analysis of 3D bioactive conformations or ensembles of potential bioactive conformations for known and novel drugs.
[1] Butler, Keith T., F. Javier Luque, and Xavier Barril. "Toward accurate relative energy predictions of the bioactive conformation of drugs." Journal of computational chemistry 30.4 (2009): 601-610.
[2] Perola, Emanuele, and Paul S. Charifson. "Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding." Journal of medicinal chemistry 47.10 (2004): 2499-2510.
[3] Tirado-Rives, Julian, and William L. Jorgensen. "Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding." Journal of medicinal chemistry 49.20 (2006): 5880-5884.
[4] Pisani, Pasquale, et al. "Describing the conformational landscape of small organic molecules through gaussian mixtures in dihedral space." Journal of Chemical Theory and Computation 10.6 (2014): 2557-2568.
[5] Juárez-Jiménez, Jordi, et al. "Assessing the Suitability of the Multilevel Strategy for the Conformational Analysis of Small Ligands." The Journal of Physical Chemistry B 119.3 (2014): 1164- 117
BCE Framework
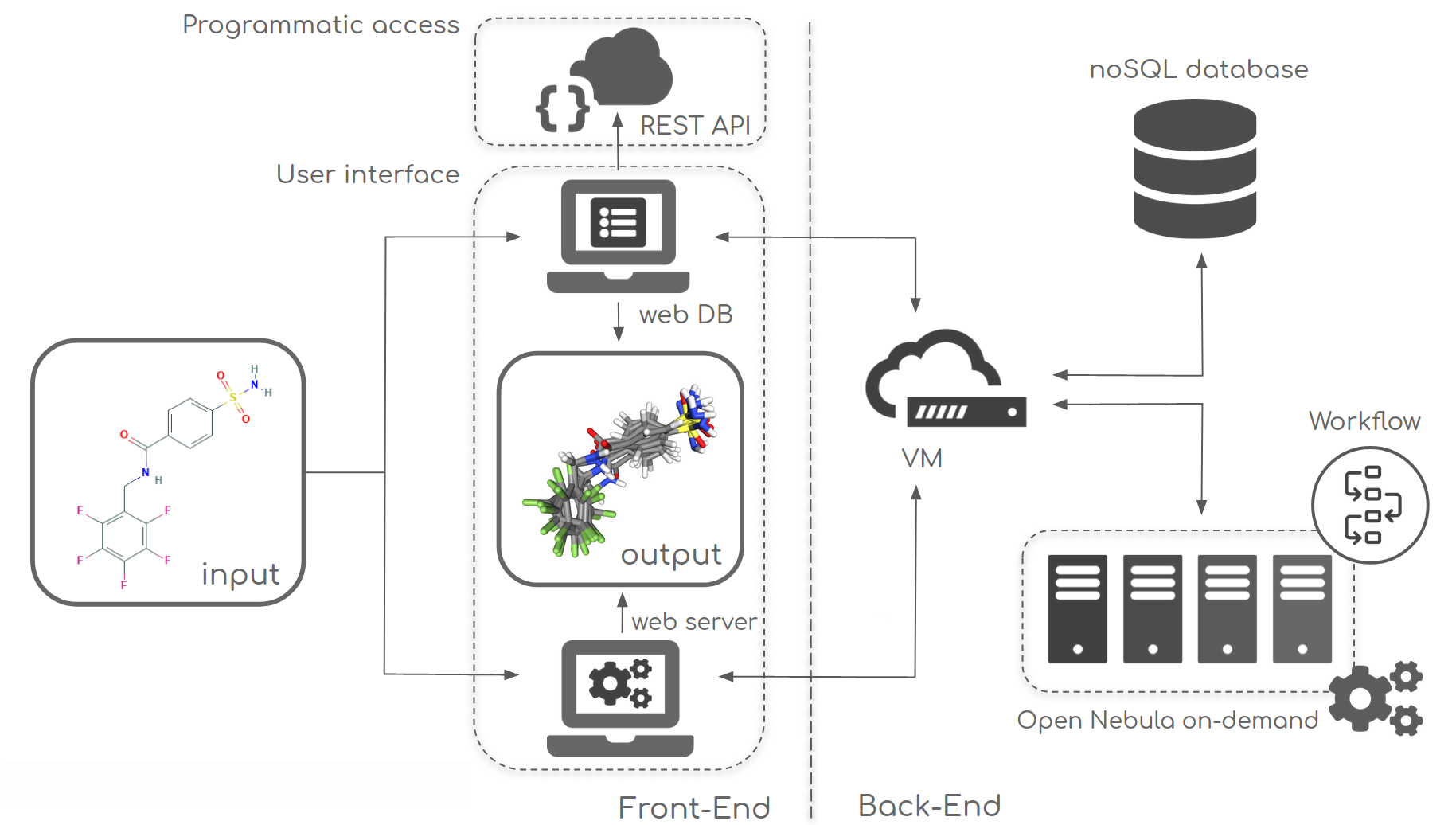
TheBioactive Conformational Ensemble (BCE) framework consists in a server from which the workflow necessary to generate representative conformers can be launched, and a database where results can be stored and made publicly available. A shared, web-based graphical interface allows the analysis of all the data.
The framework back-end is a combination of state-of-the art technology, including a Mongo noSQL database to store all generated data, and an on-demand processing model with automatic deployment of Virtual Machines (VMs) to compute the conformational ensembles.
BCE Database
The multilevel strategy was tested on more than 100 pharmaceutically relevant drug- like complexes and the whole data set with analysis is available on theBioactive Conformational Ensemble webserver. The data base is complemented with GPCR ligands, macrocycles and dual ligands (where the same ligand groups make similar interactions with related residues in both binding sites).
- Perola, Emanuele, and Paul S. Charifson. "Conformational analysis of drug- like molecules bound to proteins: an extensive study of ligand reorganization upon binding." Journal of medicinal chemistry 47.10 (2004): 2499-2510.
- Barelier, Sarah, et al. "The recognition of identical ligands by unrelated proteins." ACS chemical biology 10.12 (2015): 2772-2784.
- Shonberg, Jeremy, et al. "GPCR crystal structures: medicinal chemistry in the pocket." Bioorganic&medicinal chemistry 23.14 (2015): 3880-3906.
Bioactive Conformational Ensemble database allows you direct access to individual trajectory data and computed analyses of dihedrals, clusters, PCA, as well as analysis of relative energies obtained from QM calculations.
Programmatic access to the database is possible to facilitate high-throughput meta-analysis of the data through a REST API.
BCE web server
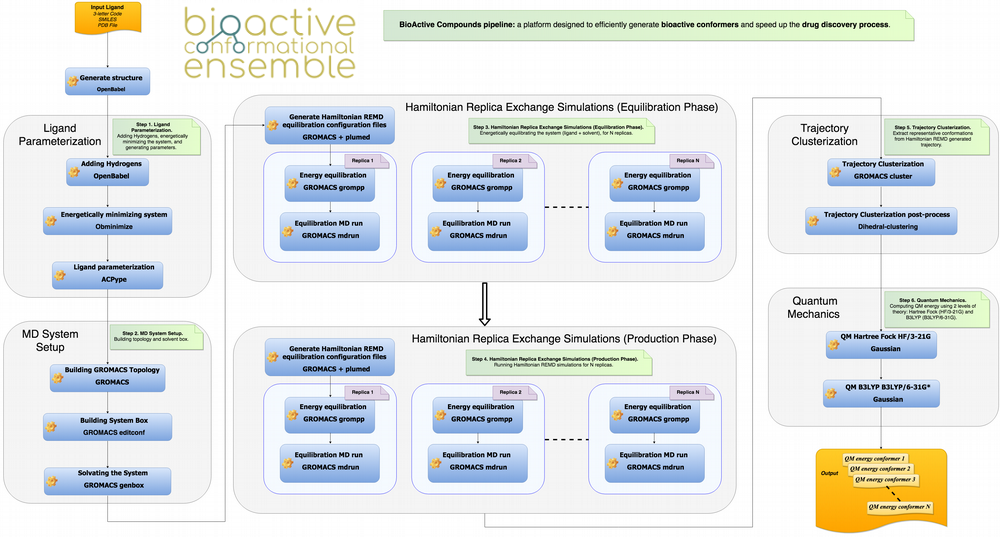
The BCE web server allows the automatic launching of a workflow intended to generate reliable conformational ensembles for a particular compound (see pipeline below). The workflow consists on a series of building blocks that generate topologies and initial structures of the molecule, define approximate force-field parameters, and derive and process classical ensembles to finally obtain representative conformations. Those conformations can be then submitted to QM calculations for further refinement. Once calculations are finished the server offers a variety of analysis on the data and the possibility to upload the results into the BCE database, where both raw data and associated analysis are accessible.