Bioactive Conformational Ensemble Help - Create New Project - Step 3: Settings
Settings
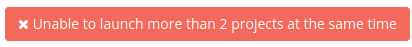
In this page users must fill in the metadata and launch the job. It is important to be aware that a user only can send two jobs at once. If you try to send more, you won't be able to send this job and the next button will be shown:
These are the sections of this page:
Project
In this section users must provide the name of the new project and optionally a description.
The non-registered users optionally can provide an e-mail address for receive a message once the job is finished. This e-mail address will only be used for this purpose. The registered users will receive this message automatically.
Inputs
In this section are shown the inputs. There are three different kind of inputs:
SMILES inputs
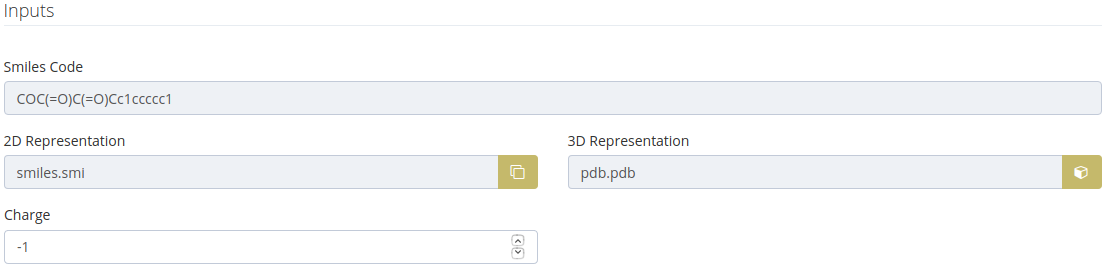
These inputs are shown when the step 1 is one of the following cases:
- Chemical Drawing: New project
- Chemical Drawing: Search by similarity in ChEMBL - EBI
- SMILES: New project
- SMILES: Search on database ChEMBL - EBI
- Browse: Search on database ChEMBL - EBI
The charge is calculated as a result of the protonation state selected.
Ligand input
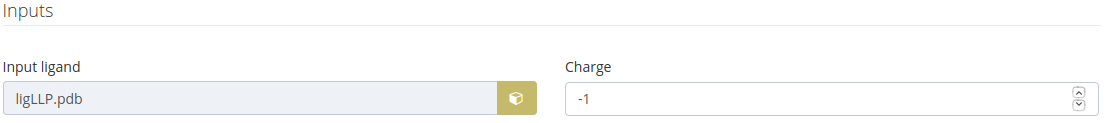
This input is shown when the step 1 is the following case:
- Upload PDB File: Ligand File
The charge is calculated as a result of the protonation state selected.
Structure + Ligand inputs
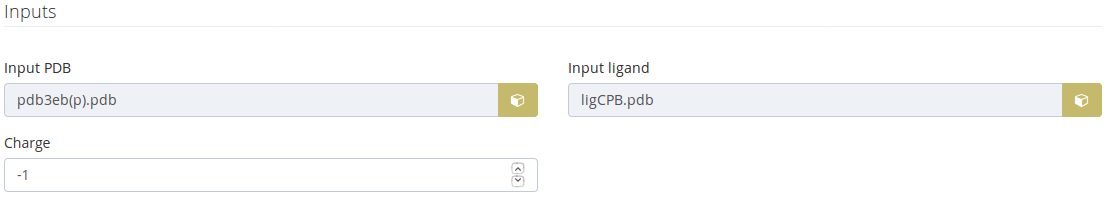
These inputs are shown when the step 1 is one of the following cases:
- Chemical Drawing: Search by similarity in Protein Data Bank
- SMILES: Search on database Protein Data Bank
- Browse: Search on database Protein Data Bank
- Upload PDB File: Structure File
- PDB ID + Ligand ID: Structure + Ligand
- PDB ID + Ligand ID: Ligand (the structure for the selected ligand is automatically uploaded)
The charge is calculated as a result of the protonation state selected.
Dihedrals
In this section, users select dihedral angles that can potentially generate symmetric conformations. Selection is done by mouse clicking on the bond formed by the two dihedral central atoms in the 3D representation of the molecule. This information will be used in the clustering process to remove symmetric conformations.
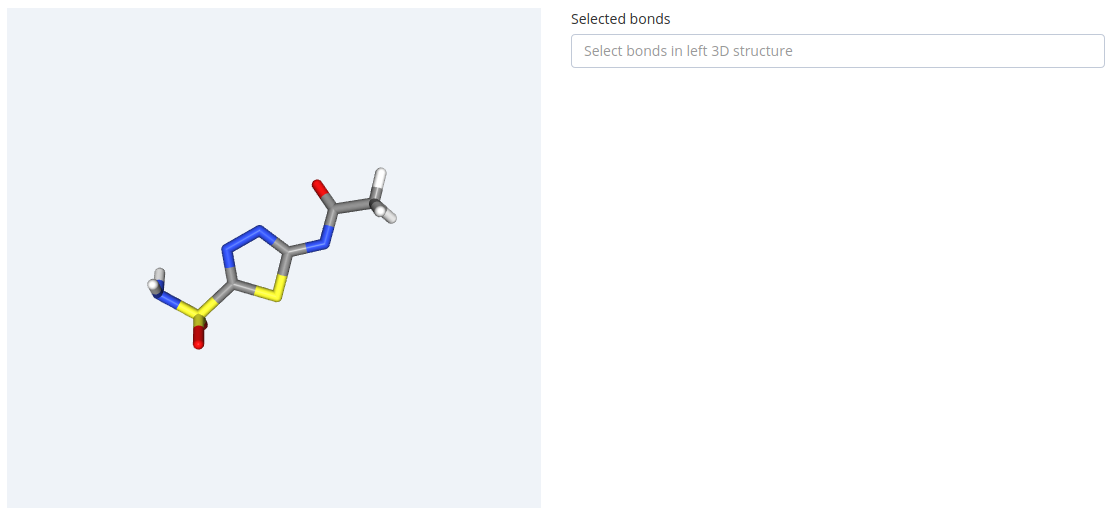
As a first step, please find the bond in the 3D structure. Passing the mouse over the bond will highlight it in brown:
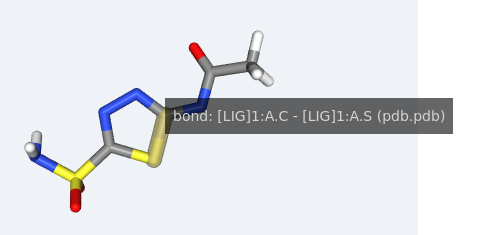
To select the bond, please click the it in the 3D structure and it will be highlighted in white and added to the drop down list on the right side of the block:
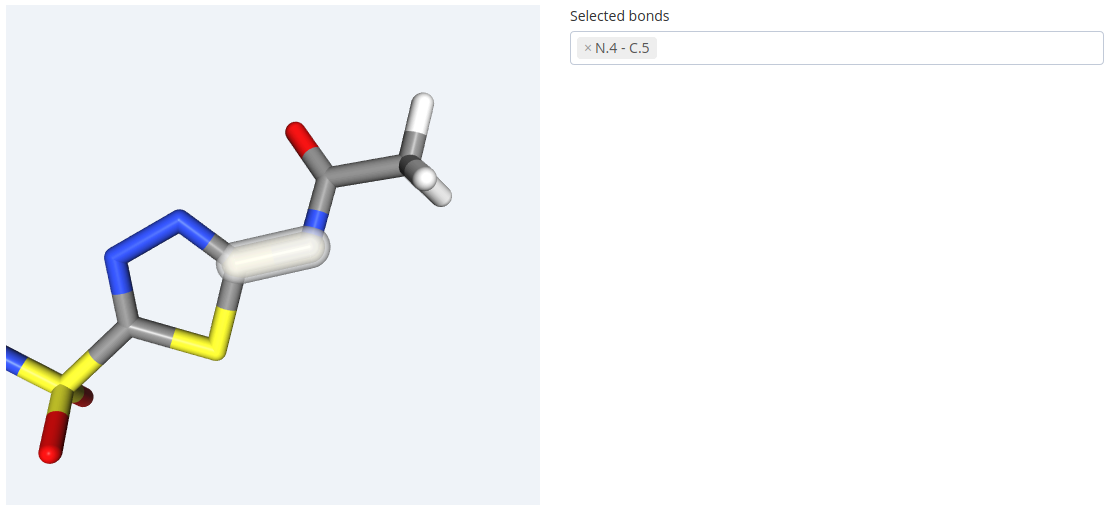
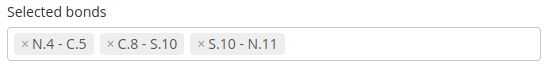
The bonds selected in the list can be removed just clicking on the cross button of each bond:
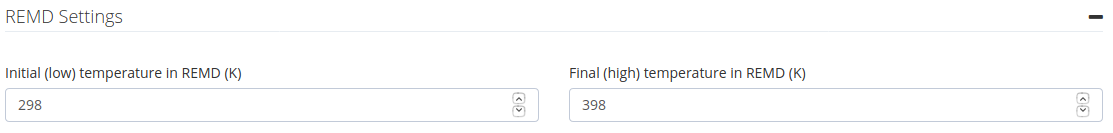
REMD Settings
Initial (low) and final (high) temperature to be used in the enhanced sampling Molecular Dynamics simulation (in K). Temperatures will be internally translated to scaling factors applied to the dihedral angles of the system in the Hamiltonian REMD algorithm.
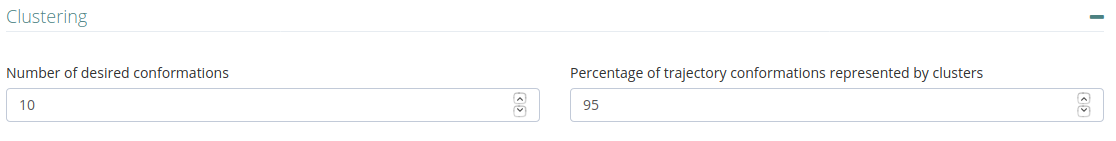
Clustering
Number of desired conformations representing a given percentage of the conformational space explored by the simulation to be kept from the trajectory in the clustering process.