Help - Outputs
Outputs
Each workflow returns its own different outputs, but the structure of these sections is common for all of them.
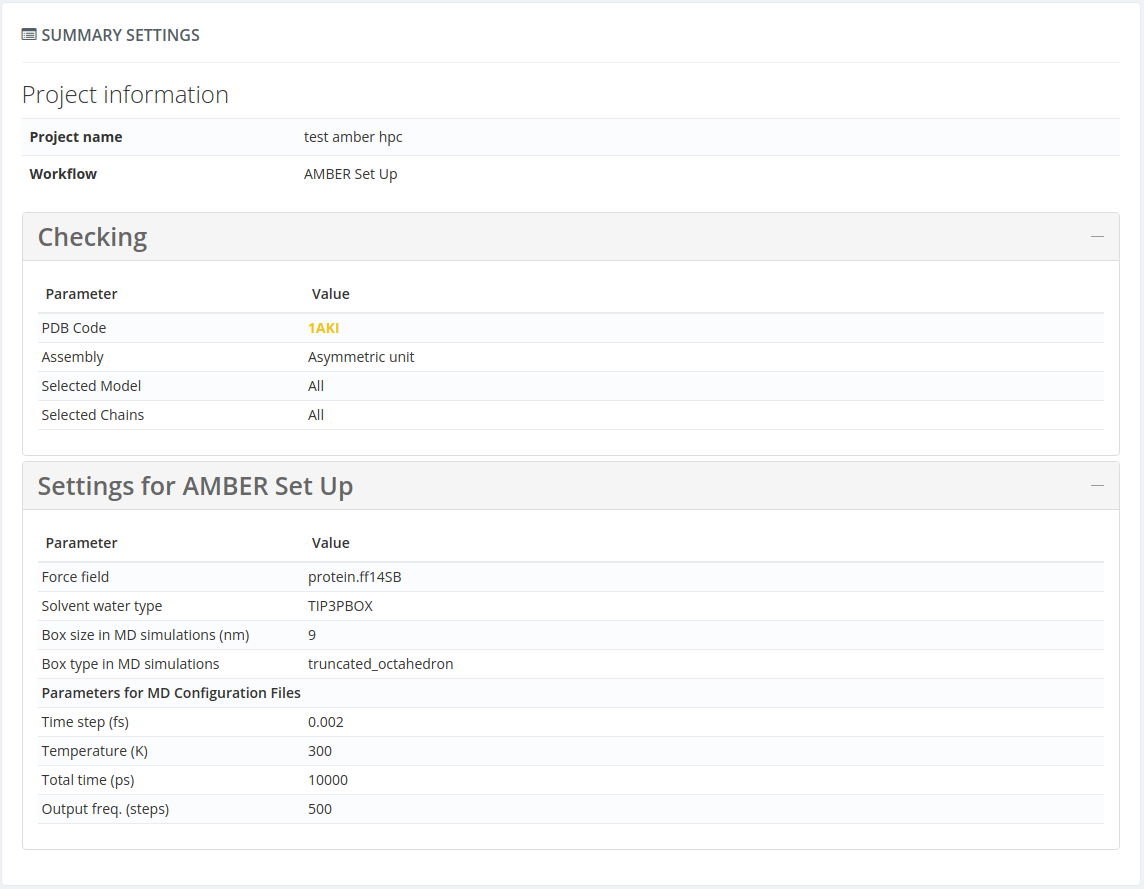
Summary settings
In this section there is a summary of all the parameters introduced by the users in the create project steps. Below there is a figure for a structure example (with checking box):
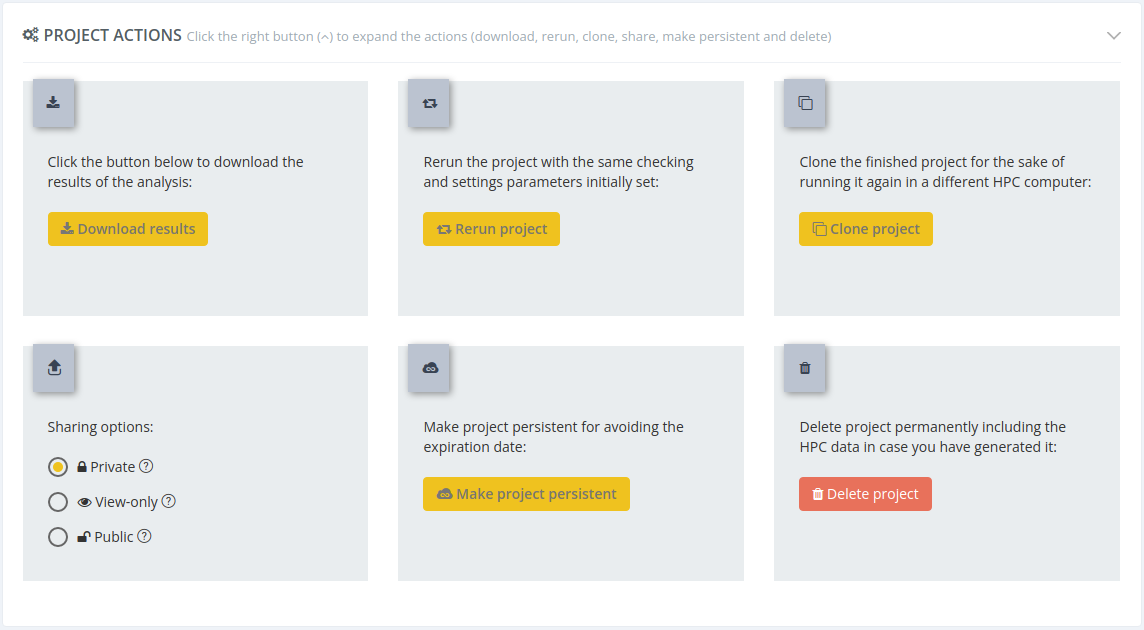
Project actions
In this section we can find all the actions related with the project:
Download results
Clicking this button all the results of the analysis will be downloaded.
Below you can find a summary for the outputs of all available workflows:
Outputs for ABC MD Setup workflow
This workflow provides a pipeline to setup DNA structures following the recommended guidelines by the Ascona B-DNA Consortium (ABC) members. It follows the work started with the NAFlex tool to offer a single, reproducible pipeline for structure preparation, ensuring reproducibility and coherence between all the members of the consortium.
Below there is a short description for all the ABC MD Setup workflow outputs:
- biobb.ABCMD.ions.parmtop: Adding additional ionic concentration.
- biobb.ABCMD.randIons.pdb: Randomly swap the positions of solvent and ions.
- biobb.ABCMD.leap.4fs.top: Generate Topology with Hydrogen Mass Partitioning (4fs).
- biobb.ABCMD.eq1.energy.dat: System Energetic Minimization, 5 Kcal/mol heavy atoms restraints (1000 cycles).
- biobb.ABCMD.eq2.energy.dat: NVT Equilibration, 5 Kcal/mol heavy atoms restraints, timestep 1fs (15ps).
- biobb.ABCMD.eq3.energy.dat: System Energetic Minimization, 2 Kcal/mol heavy atoms restraints (1000 cycles).
- biobb.ABCMD.eq4.energy.dat: System Energetic Minimization, 0.1 Kcal/mol heavy atoms restraints (1000 cycles).
- biobb.ABCMD.eq5.energy.dat: System Energetic Minimization (1000 cycles).
- biobb.ABCMD.eq6.pressure_and_density.dat: NPT Equilibration, 1 Kcal/mol heavy atoms restraints, timestep 1fs (5ps).
- biobb.ABCMD.eq7.pressure_and_density.dat: NPT Equilibration, 0.5 Kcal/mol heavy atoms restraints, timestep 1fs (5ps).
- biobb.ABCMD.eq8.pressure_and_density.dat: NPT Equilibration, 0.5 Kcal/mol backbone atoms restraints, timestep 1fs (10ps).
- biobb.ABCMD.eq9.pressure_and_density.dat: NPT Equilibration, timestep 2fs (10ps).
- biobb.ABCMD.eq10.pressure_and_density.dat: NPT Equilibration, timestep 2fs, long simulation (1ns).
- biobb.ABCMD.md.netcdf: NETCDF Free Molecular Dynamics Simulation.
- biobb.ABCMD.md.ncrst: NCRST Free Molecular Dynamics Simulation.
- biobb.ABCMD.rms_first.dat: RMSd against minimized and equilibrated snapshot.
- biobb.ABCMD.rms_exp.dat: RMSd against experimental structure.
- biobb.ABCMD.rgyr.dat: Computing Radius of Gyration to measure the protein compactness during the free MD simulation.
For more information about this workflow, please visit the workflows section in our website.
Outputs for AMBER Ligand Parameterization workflow
This workflow performs an automatic ligand parameterization for a small molecule using GAFF force field, generating parameters compatible with the AMBER MD package.
Below there is a short description for all the AMBER Ligand Parameterization workflow outputs:
- biobb.LigandParam.min.mol2: Added Hydrogen atoms to the input small molecule, according to the given pH.
- biobb.LigandParam.output.frcmod: FRCMOD ligand parameters for the input small molecule.
- biobb.LigandParam.output.inpcrd: INPCRD ligand parameters for the input small molecule.
- biobb.LigandParam.output.lib: LIB ligand parameters for the input small molecule.
- biobb.LigandParam.output.prmtop: PRMTOP ligand parameters for the input small molecule.
For more information about this workflow, please visit the workflows section in our website.
Outputs for AMBER Complex Set Up workflow
This workflow performs a simulation setup of a protein-ligand(s) complex system, compatible with the AMBER MD package.
Below there is a short description for all the AMBER Complex Set Up workflow outputs:
- biobb.AMBERMDsetup.leap.top: AMBER topology corresponding to the protein structure.
- biobb.AMBERMDsetup.h_min.dat: Checking hydrogen minimization results.
- biobb.AMBERMDsetup.n_min.dat: Checking energy minimization results.
- biobb.AMBERMDsetup.ions.parmtop: PARMTOP result of neutralizing the system and adding an additional ionic concentration.
- biobb.AMBERMDsetup.ions.pdb: PDB result of neutralizing the system and adding an additional ionic concentration.
- biobb.AMBERMDsetup.min.dat: Checking energy minimization results.
- biobb.AMBERMDsetup.temp.dat: Checking system warming up.
- biobb.AMBERMDsetup.nvt.dat: Checking NVT Equilibration results.
- biobb.AMBERMDsetup.npt.dat: Checking NPT Equilibration results.
- biobb.AMBERMDsetup.free.netcdf: Portable binary run file to run a free MD simulation (NETCDF).
- biobb.AMBERMDsetup.free.rst: Portable binary run file to run a free MD simulation (RST).
- biobb.AMBERMDsetup.rms_first.dat: RMSd against minimized and equilibrated snapshot.
- biobb.AMBERMDsetup.rms_exp.dat: RMSd against experimental structure.
- biobb.AMBERMDsetup.rgyr.dat: Computing Radius of Gyration to measure the protein compactness during the free MD simulation.
For more information about this workflow, please visit the workflows section in our website.
Outputs for AMBER Set Up workflow
This workflow performs a simulation setup of a protein system, compatible with the AMBER MD package.
Below there is a short description for all the AMBER Set Up workflow outputs:
- biobb.AMBERMDsetup.leap.top: AMBER topology corresponding to the protein structure.
- biobb.AMBERMDsetup.h_min.dat: Checking hydrogen minimization results.
- biobb.AMBERMDsetup.n_min.dat: Checking energy minimization results.
- biobb.AMBERMDsetup.ions.parmtop: PARMTOP result of neutralizing the system and adding an additional ionic concentration.
- biobb.AMBERMDsetup.ions.pdb: PDB result of neutralizing the system and adding an additional ionic concentration .
- biobb.AMBERMDsetup.min.dat: Checking energy minimization results.
- biobb.AMBERMDsetup.temp.dat: Checking system warming up.
- biobb.AMBERMDsetup.nvt.dat: Checking NVT Equilibration results.
- biobb.AMBERMDsetup.npt.dat: Checking NPT Equilibration results.
- biobb.AMBERMDsetup.free.netcdf: Portable binary run file to run a free MD simulation (NETCDF).
- biobb.AMBERMDsetup.free.rst: Portable binary run file to run a free MD simulation (RST).
- biobb.AMBERMDsetup.rms_first.dat: RMSd against minimized and equilibrated snapshot.
- biobb.AMBERMDsetup.rms_exp.dat: RMSd against experimental structure.
- biobb.AMBERMDsetup.rgyr.dat: Computing Radius of Gyration to measure the protein compactness during the free MD simulation.
For more information about this workflow, please visit the workflows section in our website.
Outputs for CNS/XPLOR Ligand Parameterization workflow
This workflow performs an automatic ligand parameterization for a small molecule using GAFF force field, generating parameters compatible with the CNS/XPLOR MD package.
Below there is a short description for all the CNS/XPLOR Ligand Parameterization workflow outputs:
- biobb.LigandParam.min.mol2: Added Hydrogen atoms to the input small molecule, according to the given pH.
- biobb.LigandParam.output.par: PAR ligand parameters for the input small molecule.
- biobb.LigandParam.output.inp: INP ligand parameters for the input small molecule.
- biobb.LigandParam.output.top: TOP ligand parameters for the input small molecule.
- biobb.LigandParam.output.pdb: PDB ligand parameters for the input small molecule.
For more information about this workflow, please visit the workflows section in our website.
Outputs for GROMACS Complex Set Up workflow
This workflow performs a simulation setup of a protein-ligand complex system, compatible with the GROMACS MD package.
Below there is a short description for all the GROMACS Complex Set Up workflow outputs:
- biobb.ComplexMDsetup.gro: Protein system topology.
- biobb.ComplexMDsetup.H.pdb: Protein-ligand complex PDB file with hydrogens.
- biobb.ComplexMDsetup.genion.zip: Replace solvent molecules with ions to neutralize the system.
- biobb.ComplexMDsetup.min.energy.xvg: Checking Energy Minimization results.
- biobb.ComplexMDsetup.nvt.temp.xvg: Checking NVT Equilibration results.
- biobb.ComplexMDsetup.npt.PD.xvg: Checking NPT Equilibration results.
- biobb.ComplexMDsetup.rmsd_first.xvg: RMSd against minimized and equilibrated snapshot.
- biobb.ComplexMDsetup.rmsd_exp.xvg: RMSd against experimental structure.
- biobb.ComplexMDsetup.rgyr.xvg: Computing Radius of Gyration to measure the protein compactness during the free MD simulation .
- biobb.ComplexMDsetup.gro: Removing water molecules and ions from the resulting structure.
- biobb.ComplexMDsetup.tpr: Portable binary run file to run a long MD (production) simulation.
For more information about this workflow, please visit the workflows section in our website.
Outputs for DNA helical parameters workflow
This tutorial aims to illustrate the process of extracting structural and dynamical properties from a DNA MD trajectory helical parameters, step by step, using the BioExcel Building Blocks library (biobb). The particular example used is the Drew Dickerson Dodecamer sequence -CGCGAATTCGCG- (PDB code 1BNA). The trajectory used is a 500ns-long MD simulation taken from the BigNASim database (NAFlex_DDD_II entry).
Below there is a short description for all the DNA helical parameters workflow outputs:
- biobb.DNAHelparms.canal.zip: Time series (series property) and histograms (histo property) for the parameter variations during dynamics.
- biobb.DNAHelparms.bps.averages.zip: Helical Base Pair Step (Inter Base Pair) Parameters.
- biobb.DNAHelparms.bp.averages.zip: Helical Base Pair (Intra Base Pair) Parameters.
- biobb.DNAHelparms.abp.averages.zip: Axis Base Pair Parameters.
- biobb.DNAHelparms.grv.averages.zip: DNA Grooves averages.
- biobb.DNAHelparms.pck.averages.zip: Sugar Puckering annotations.
- biobb.DNAHelparms.ag.averages.zip: Rotations around alpha/gamma torsions.
- biobb.DNAHelparms.bIbII.averages.zip: The concerted rotation around zeta/epsilon torsions generates two major conformers: BI and BII.
- biobb.DNAHelparms.bps.timeseries.zip: Helical Base Pair Step (Inter-Base Pair) Helical Parameters.
- biobb.DNAHelparms.bp.timeseries.zip: Helical Base Pair (Intra-Base Pair) Helical Parameters.
- biobb.DNAHelparms.abp.timeseries.zip: Axis Base Pair Parameters.
- biobb.DNAHelparms.grv.timeseries.zip: DNA Grooves parameters.
- biobb.DNAHelparms.bt.timeseries.zip: Backbone Torsions parameters.
- biobb.DNAHelparms.basepair_stiffness.zip: Basepair stiffness.
- biobb.DNAHelparms.bimodality.zip: DNA bimodality.
- biobb.DNAHelparms.intraseqcorr.zip: Sequence Correlations: Intra-base pairs.
- biobb.DNAHelparms.interseqcorr.zip: Sequence Correlations: Inter-base pair steps.
- biobb.DNAHelparms.intrahpcorr.zip: Helical Parameter Correlations: Intra-base pairs.
- biobb.DNAHelparms.interhpcorr.zip: Helical Parameter Correlations: Inter-base pair steps.
- biobb.DNAHelparms.intrabpcorr.zip: Neighboring steps Correlations: Intra-base pairs.
- biobb.DNAHelparms.interbpcorr.zip: Neighboring steps Correlations: Inter-base pair steps.
For more information about this workflow, please visit the workflows section in our website.
Outputs for Docking Autodock Vina workflow
This workflow performs the process of protein-ligand docking, step by step, using the BioExcel Building Blocks library (biobb).
Below there is a short description for all the Docking Autodock Vina workflow outputs:
- biobb.DockingVina.pocket.pqr: The selected protein cavity (pocket) from the fpocket results.
- biobb.DockingVina.box.pdb: The box surrounding the selected protein cavity (pocket), to be used in the docking procedure.
- biobb.DockingVina.ligand.pdbqt: The small molecule structure prepared for the docking procedure.
- biobb.DockingVina.receptor.pdbqt: The target protein structure prepared for the docking procedure.
- biobb.DockingVina.poses.pdbqt: Docking poses for the ligand generated by AutoDock Vina
- biobb.DockingVina.output.log: AutoDock Vina log.
- biobb.DockingVina.output.pdb: Best ligand pose converted to PDB format
For more information about this workflow, please visit the workflows section in our website.
Outputs for GROMACS Ligand Parameterization workflow
This workflow performs an automatic ligand parameterization for a small molecule using GAFF force field, generating parameters compatible with the GROMACS MD package.
Below there is a short description for all the GROMACS Ligand Parameterization workflow outputs:
- biobb.LigandParam.min.mol2: Added Hydrogen atoms to the input small molecule, according to the given pH.
- biobb.LigandParam.output.gro: GRO ligand parameters for the input small molecule.
- biobb.LigandParam.output.itp: ITP ligand parameters for the input small molecule.
- biobb.LigandParam.output.top: TOP ligand parameters for the input small molecule.
For more information about this workflow, please visit the workflows section in our website.
Outputs for GROMACS OPLS/AA Ligand Parameterization workflow
This workflow performs an automatic ligand parameterization for a small molecule using GAFF force field, generating parameters compatible with the GROMACS MD package and the OPLS/AA force field.
Below there is a short description for all the GROMACS OPLS/AA Ligand Parameterization workflow outputs:
- biobb.LigandParam.min.mol2: Added Hydrogen atoms to the input small molecule, according to the given pH.
- biobb.LigandParam.output.itp: ITP ligand parameters for the input small molecule.
- biobb.LigandParam.output.top: TOP ligand parameters for the input small molecule.
For more information about this workflow, please visit the workflows section in our website.
Outputs for Protein MD Analysis workflow
TODO
Below there is a short description for all the Protein MD Analysis workflow outputs:
- biobb.MDProtein.leap.average.pdb: Trajectory structure average
- biobb.MDProtein.rmsf.dat: RMSd (against 1st snp.) plot for the trajectory
- biobb.MDProtein.rmsav.dat: RMSd (against all frames) plot for the trajectory
- biobb.MDProtein.bfactor.dat: Trajectory Bfactor fluctuations plot
- biobb.MDProtein.bfactor.pdb: Trajectory Bfactor fluctuations structure
- biobb.MDProtein.rgyr.dat: Radius of Gyration plot for the trajectory
- biobb.MDProtein.traj.trr: Trajectory GMX compatible format
- biobb.MDProtein.cluster.pdb: Clustering structures from the trajectory
- biobb.MDProtein.cluster.log: Clustering log file
For more information about this workflow, please visit the workflows section in our website.
Outputs for GROMACS Set Up workflow
This workflow performs the process of setting up a simulation system containing a protein, step by step, using the GROMACS tools of the BioExcel Building Blocks library (biobb).
Below there is a short description for all the GROMACS Set Up workflow outputs:
- biobb.MDsetup.top.zip: Replace solvent molecules with ions to neutralize the system.
- biobb.MDsetup.tpr: Portable binary run file to run a free MD simulation.
- biobb.MDsetup.gro: GRO for short free MD simulation.
- biobb.MDsetup.cpt: CPT for short free MD simulation.
- biobb.MDsetup.xtc: Imaged MD trajectory.
- biobb.MDsetup.top.pdb: Output dry structure.
- biobb.MDsetup.energy.xvg: Checking Energy Minimization results.
- biobb.MDsetup.rgyr.xvg: Radius of Gyration to measure the protein compactness during the free MD simulation .
- biobb.MDsetup.rmsd_first.xvg: RMSd against minimized and equilibrated snapshot (backbone atoms).
- biobb.MDsetup.rmsd_exp.xvg: RMSd against experimental structure (backbone atoms).
- biobb.MDsetup.prod.tpr: Portable binary run file to run a long MD (production) simulation.
For more information about this workflow, please visit the workflows section in our website.
Outputs for GROMACS Set Up workflow
This workflow performs a simulation setup of a protein system, compatible with the GROMACS MD package.
Below there is a short description for all the GROMACS Set Up workflow outputs:
- biobb.MDsetup.top.zip: Replace solvent molecules with ions to neutralize the system.
- biobb.MDsetup.tpr: Portable binary run file to run a free MD simulation.
- biobb.MDsetup.gro: GRO for short free MD simulation.
- biobb.MDsetup.cpt: CPT for short free MD simulation.
- biobb.MDsetup.xtc: Imaged MD trajectory.
- biobb.MDsetup.top.pdb: Output dry structure.
- biobb.MDsetup.energy.xvg: Checking Energy Minimization results.
- biobb.MDsetup.rgyr.xvg: Radius of Gyration to measure the protein compactness during the free MD simulation .
- biobb.MDsetup.rmsd_first.xvg: RMSd against minimized and equilibrated snapshot (backbone atoms).
- biobb.MDsetup.rmsd_exp.xvg: RMSd against experimental structure (backbone atoms).
- biobb.MDsetup.prod.tpr: Portable binary run file to run a long MD (production) simulation.
For more information about this workflow, please visit the workflows section in our website.
Rerun project
Clicking this button reruns the project with the same checking and settings parameters initially set.
Clone project
Clicking this button clones the finished project for the sake of running it again in a different HPC computer.
Sharing options
This menu allows to change the project permissions:
- Private: Only the owner can access this project through his or her private area
- View-only: The project is visible to everybody that have the link
- Public: All the users with the link can fork it
Make project persistent
Clicking this button makes the project persistent to avoid the expiration date.
Delete project
Clicking this button deletes the project permanently including the HPC data in case it has been generated.
Analysis results
In this section, the outputs of the workflow are shown. Each workflow has its own kind of output data: static 3D structure, topology + trajectory, plots, tables, and so on. Below there is a figure for a small ligand example:
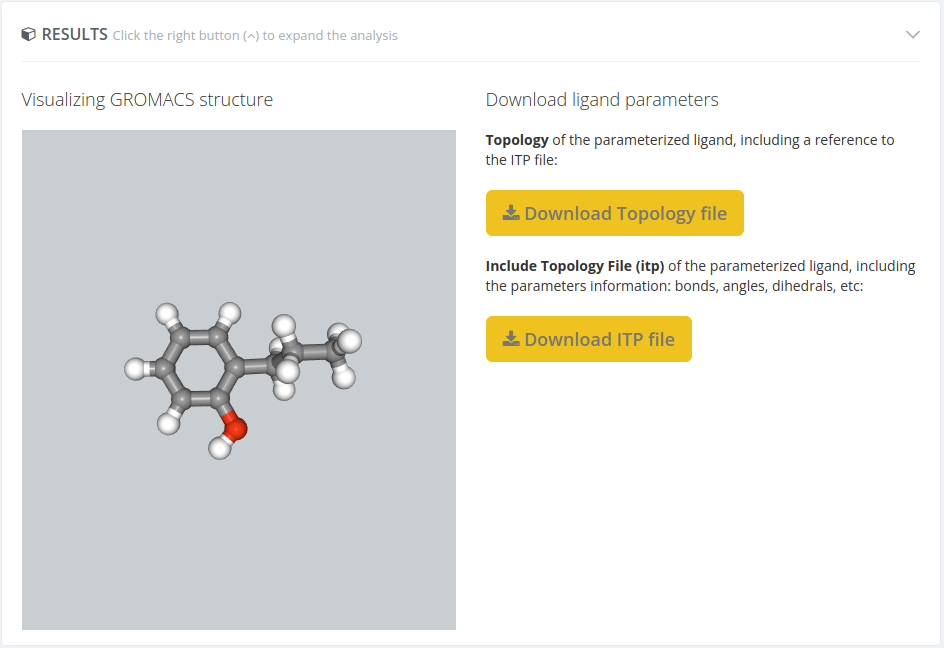
Note that all workflows have different results.
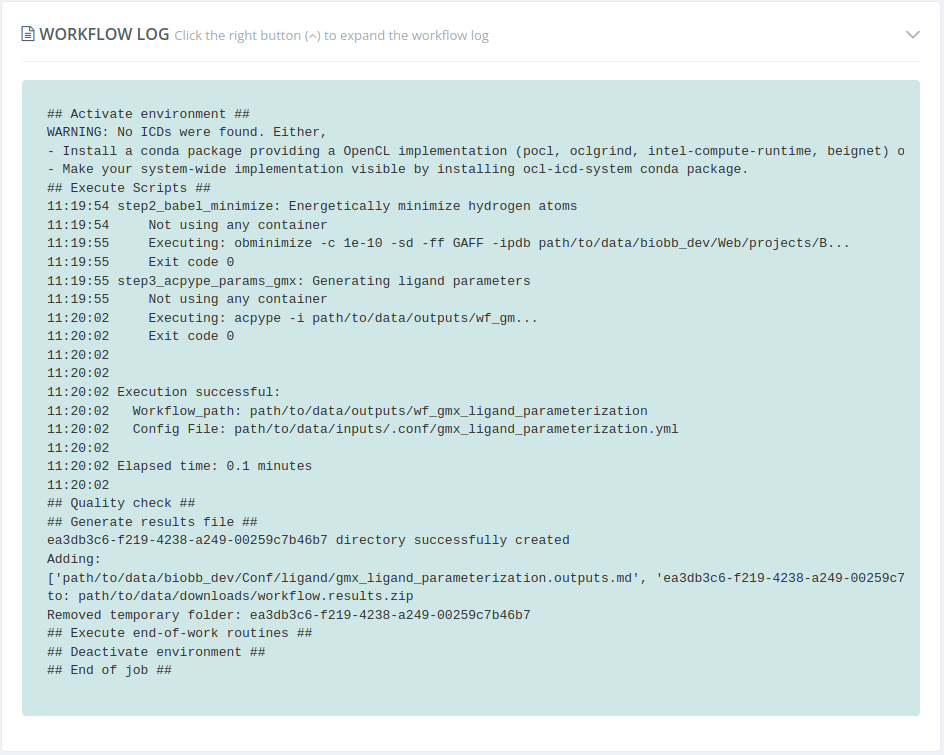
Workflow log
Tn this section, the internal workflow log is shown . This log is especially useful in case of workflow error:
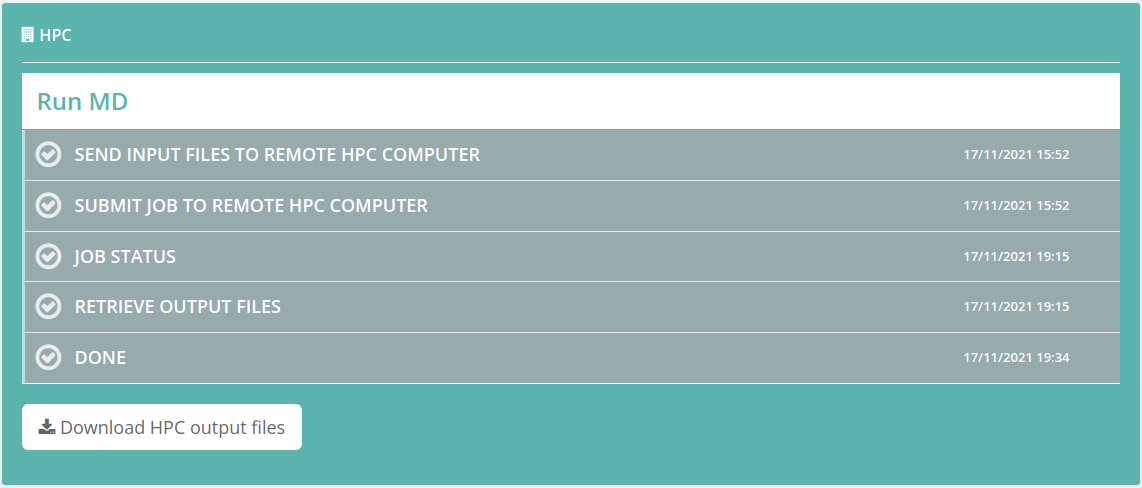
HPC
In some workflows, there is the option of preparing configuration files to run a long MD simulation in users own premises. That means that some data generated by the Workflow can be sent to a HPC supercomputer and then run there a job with this data and retrieve the outputs again to the BioBB Workflows web.
At this moment, BioBB Workflows website offers the following supercomputers for running jobs from this website:
Hence, all users with an account in these supercomputers can launch production jobs from this website.
Once the job is launched to a HPC computer, all the process is automated until the end:
- Send input files from BioBB Workflows to the user account of the HPC Computer
- Submit Job to the HPC Computer
- Check Job Status (submitted, running, finishing…)
- Retrieve of output files to your personal space in BioBB Workflows
Once the process is finished, the files generated remotely in the HPC Computer are available for downloading from the outputs page of BioBB Workflows, though of course they will also be available in the user HPC Computer account.
The process to configure a supercomputer account in BioBB Workflows is explained in My profile section.