Help - Ascona B-DNA consortium Tutorial
Ascona B-DNA consortium Tutorial
Since the discovery of the double helix, DNA structure has been a central topic in biology, chemistry and physics. Recent studies have revealed how flexible and structurally polymorphic DNA structure can be, with physical properties defining a secondary genetic code that modulates the accessibility to effector proteins. Sequence-dependent structural properties of DNA are now considered crucial to rationalize how proteins and small molecules recognize DNA and modulate its activity. The Ascona B-DNA consortium (ABC), created in 2004 to coordinate a community effort for characterizing the sequence-dependent physical properties of DNA by means of accurate atomistic MD simulations, have published a collection of scientific studies directly contributing to advances on DNA force-fields, coarse grained and mesoscopic models of DNA. One of the most important ABC projects involved the analysis of all existing tetra nucleotides by means of atomistic MD simulations (miniABC), developing and sharing standards for the simulation protocols and DNA flexibility analysis. BioBB Workflows can be used to reproduce the pipeline used by ABC to model the DNA structure, prepare the MD system, run the simulation, and finally extract flexibility patterns (sequence-dependent structural properties) from the resulting trajectory.
ABC MD Setup
The first workflow chosen for this study is the ABC MD Setup Pipeline, which reproduces the protocol built by the ABC members to prepare a DNA system. The input sequence is one of the 13 sequences used in the miniABC study: 5’-GCAACGTGCTATGGAAGC-3’. The sequence is introduced in the Provide DNA/RNA Seq. step, and the canonical right-handed B-DNA structure (Chandrasekaran and Arnott. J Biomol Struct Dyn, 1996) is selected:
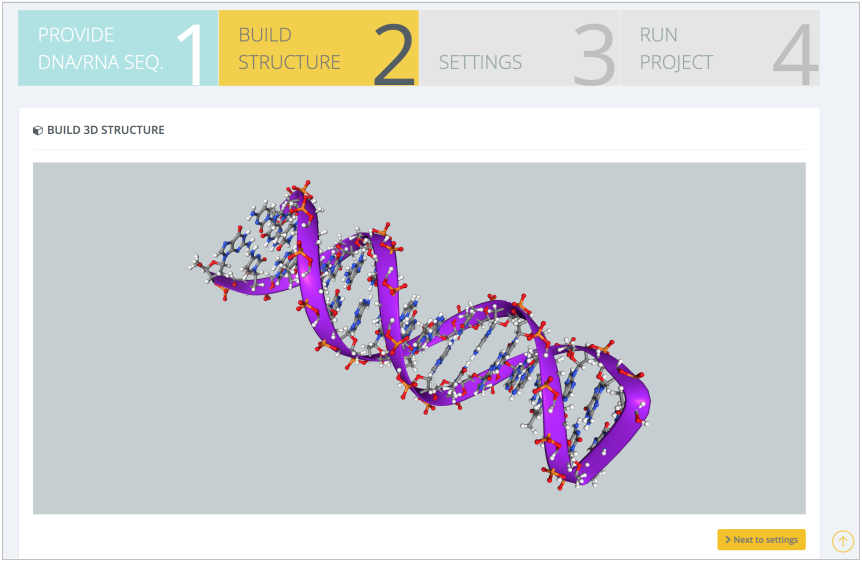
The server will then internally model the structure and display it in the Build Structure intermediate step:
In the next step, Settings, the most important workflow parameters can be modified:
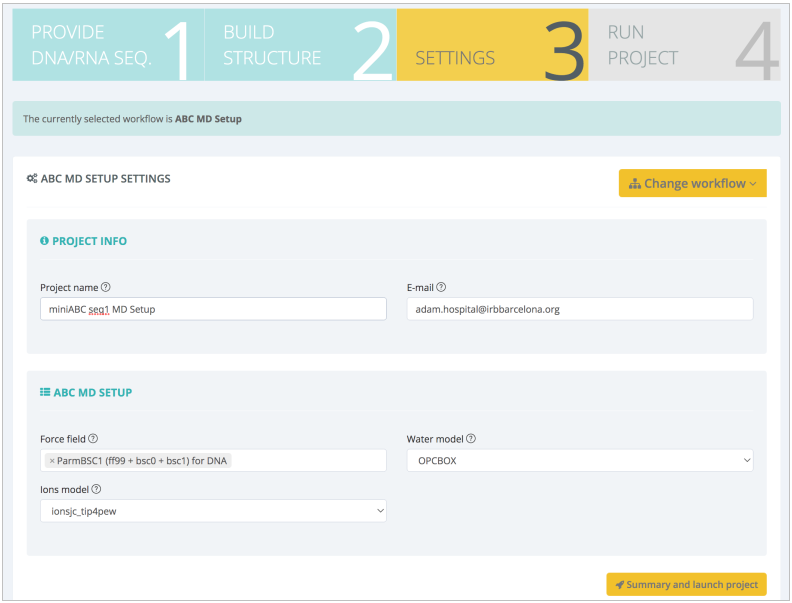
In this case, different force field, water and ions models can be selected. Default values are taken from the ABC work (ParmBSC1, Ivani et al. Nature Methods, 2016, OPC Izadi et al. J Phys Chem Lett, 2014 and Joung and Cheatham. J Phys Chem B, 2008). After checking a summary of the most important data that is going to be used in the workflow (sequence, structure, settings), the workflow can be launched, either downloading the Python/CWL scripts and running it locally or executing it in the same web platform :
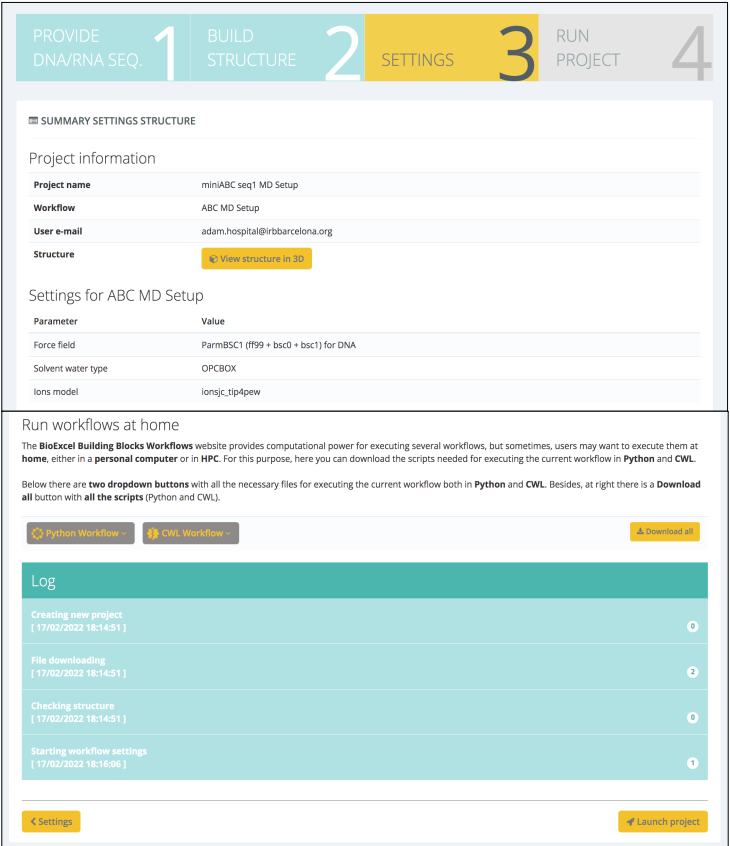
When the workflow is launched in the server, a step-by-step progress is shown:
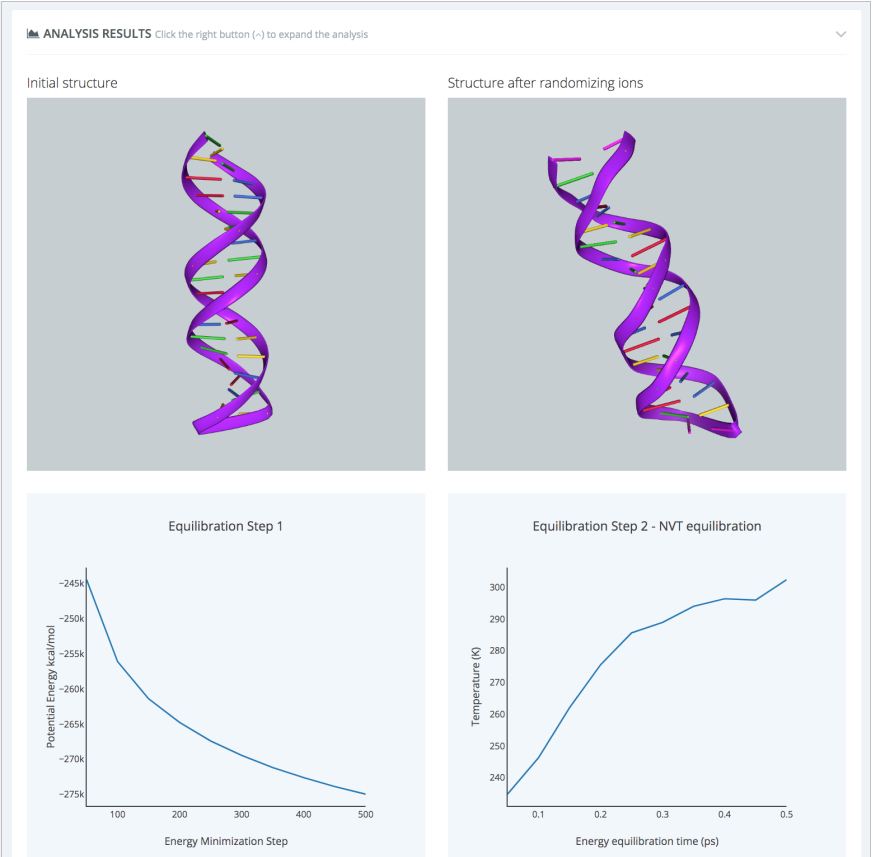
When finished, results are presented in a workflow-specific interface. In this case, changes in the system energy values during the minimization and equilibration steps are shown, giving a quick idea on the consistency of the setup process:
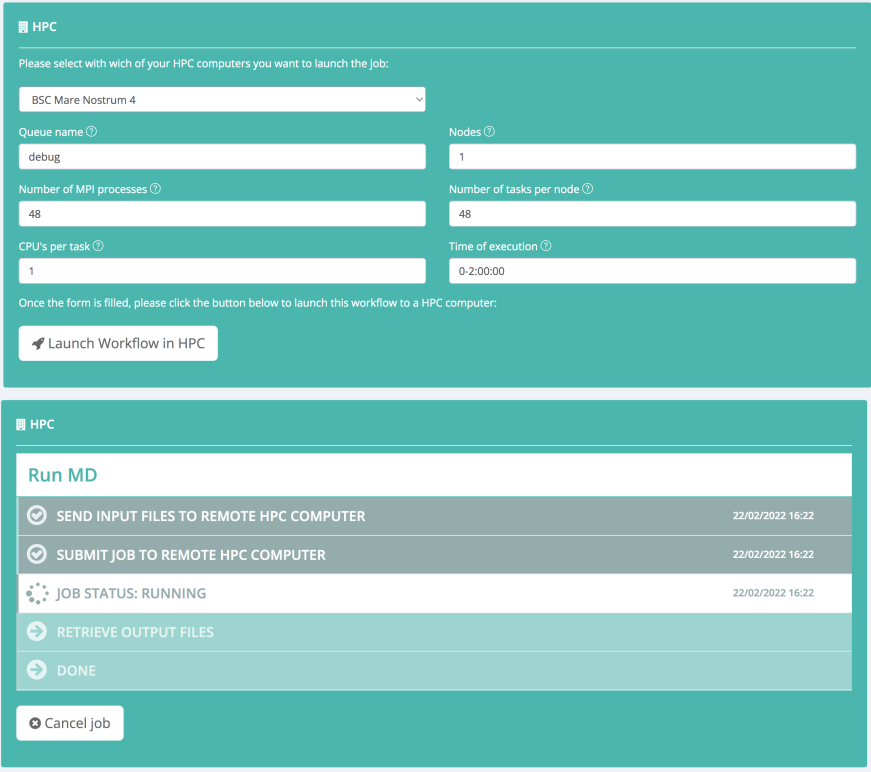
Final results can be then downloaded and used as inputs for the production MD simulation. Alternatively, the production MD can be remotely launched to a BioBB Workflows compatible HPC cluster:
Structural DNA Helical Parameters
After running the production MD simulation and processing the obtained trajectory (reimaging, dehydrating), the server can be used again to calculate sequence-dependent structural properties. The workflow Structural DNA Helical Parameters takes as input a pair of MD files: topology and coordinates:
The trajectory is loaded into the server and shown for visual inspection, together with sequence strands nucleotide information, basic for the helical parameters study:
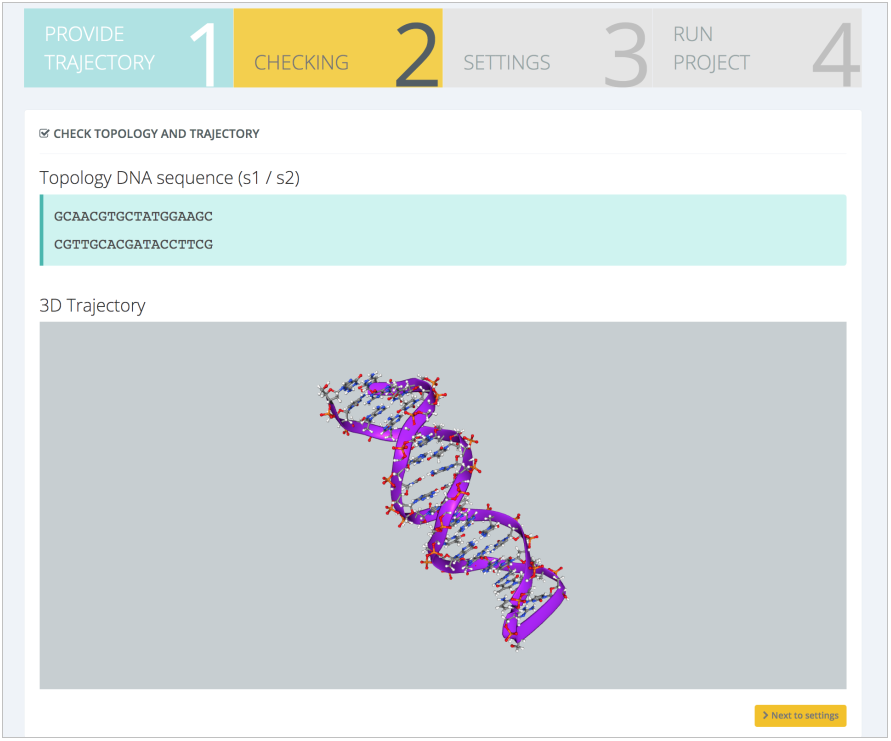
This information is automatically added as default values for the workflow inputs:
After launching the workflow and waiting for the completion of all the steps:
A new workflow-specific interface will be opened, with the flexibility information extracted from the trajectory:
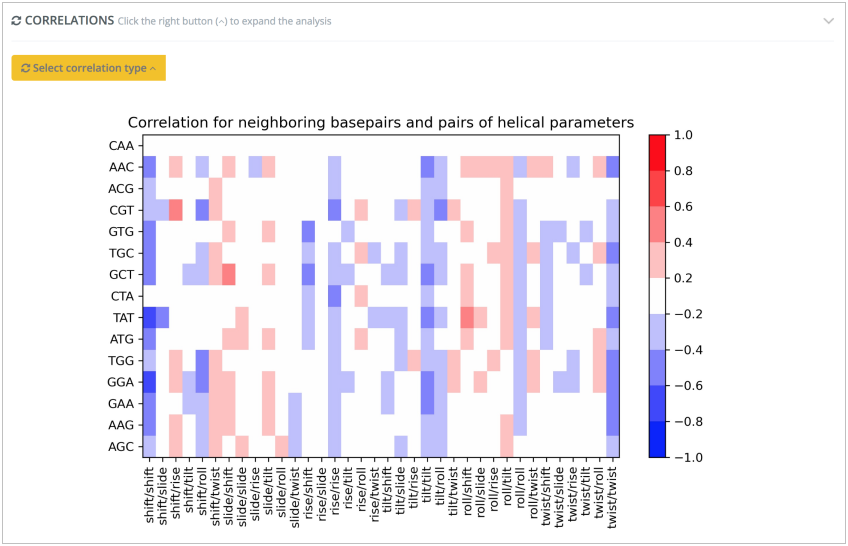
Helical parameters are computed and shown in 2D plots along and averaged over time:

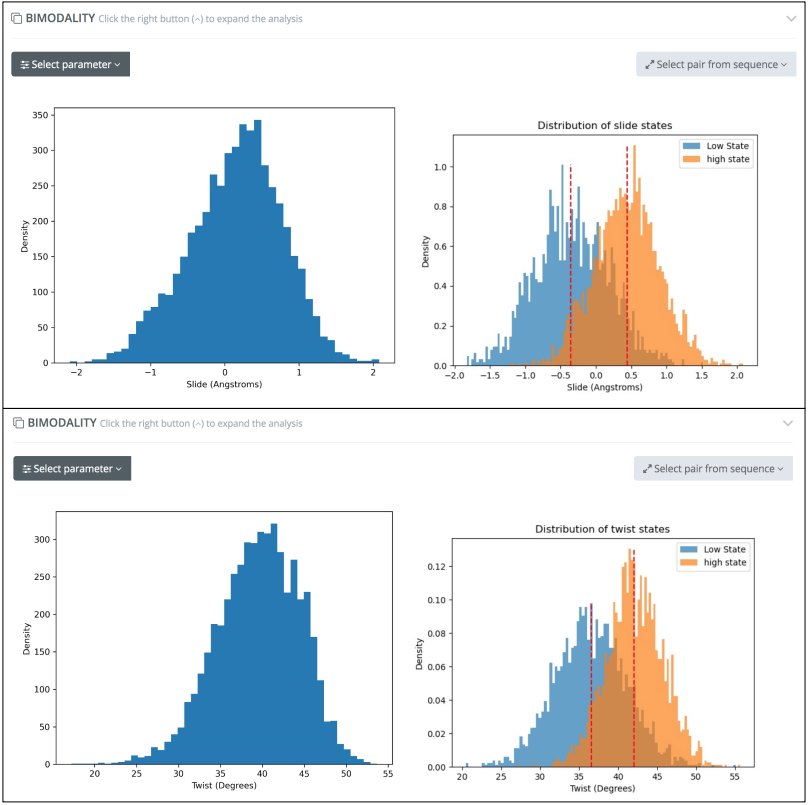
Bimodality analysis on top of base pair step helical parameters allows an easy identification of non-Gaussian distributions such as the high/low Slide values in CTAT tetramers, or the high/low Twist values in ACGT tetramers:
Reproducing results presented in Dans et al., Nucleic Acids Res, 2014 and in Balaceanu et al., Nucleic Acids Res, 2019. Correlations between helical base pair step parameters are also computed, reproducing the negative correlation values found for roll-twist parameters in RR (purine-purine) and RY (purine-pyrimidine) steps and the positive correlations found for shift-tilt parameters in RR steps, as shown in Dans et al., Nucleic Acids Res, 2019: