Help - SARS-CoV-2 - hACE2 Tutorial
SARS-CoV-2 - hACE2 Tutorial
The complex between the SARS-CoV-2 Receptor Binding Domain (RBD) with the human Angiotensin Converting Enzyme 2 (hACE2) is one of the most studied protein complex of the present time. Mutations on the viral RBD domain have been extensively studied to understand changes in the infectivity level of the SARS-CoV-2 virus from the different strain variants. As an example, the alpha variant (B.1.1.7), also known as the British variant, was estimated to be 40–80% more transmissible than the wild-type SARS-CoV-2. This variant involved a key mutation in the Receptor Binding Domain of the viral Spike protein: N501Y. BioBB Workflows can be used to model the mutation and prepare MD simulations for both the Wild Type and the mutated RBD domains in complex with the human ACE2 protein, in order to investigate the electrostatic and interaction pattern changes induced by the mutation.
The workflow chosen for this study is the GROMACS Protein MD Setup with Mutations, and the input structure is the novel coronavirus spike RBD domain complexed with its receptor ACE2 (PDB code 6LZG, Wang et al. Cell, 2020). The PDB structure is found and selected through the PDB databank browse section of the Provide Structure step:
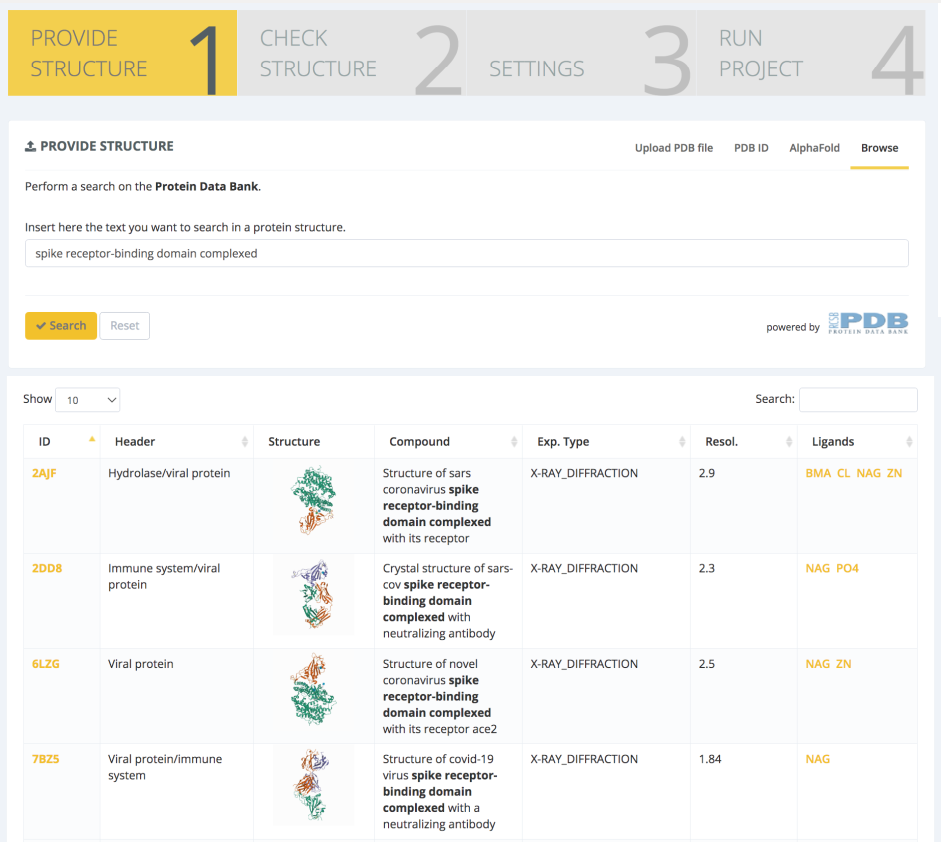
An exhaustive structure checking is then launched, with results being presented in the Structure Checking step:
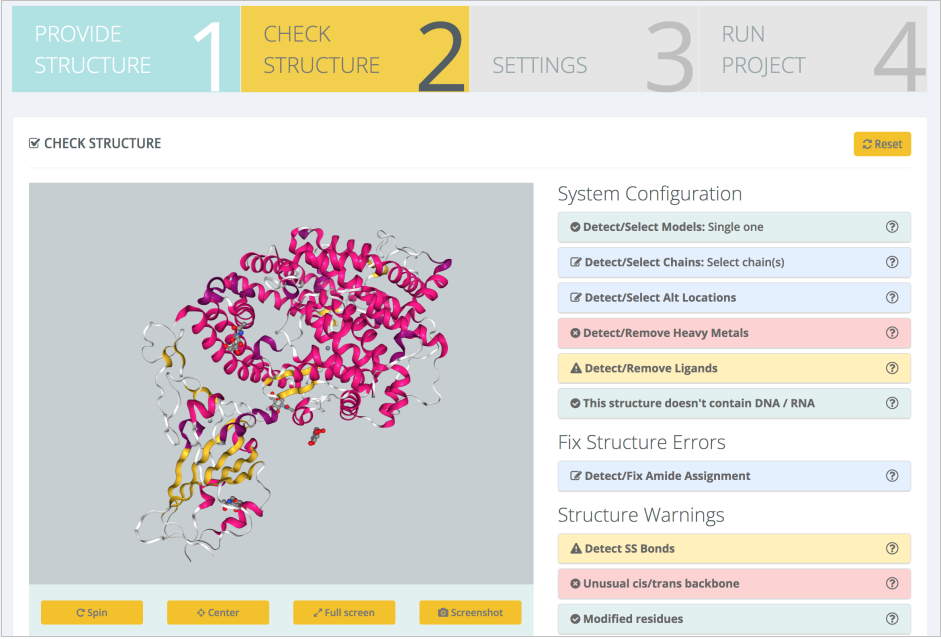
Moving to the Settings step, the interface offers the possibility to model interesting residue mutations:
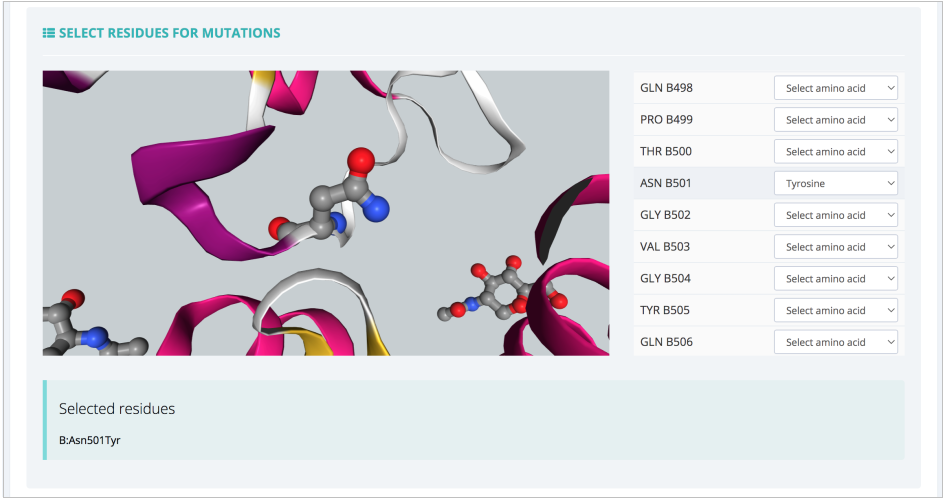
The list of residues is shown together with a 3D representation, which helps in finding the right spot to modify and study. In this particular example, ASN501 from the RBD protein is mutated to a Tyrosine residue, reproducing thus the key mutation experimentally known to appear in the SARS-CoV-2 alpha variant. In the same Settings step, the most important parameters for the MD setup workflow can also be modified:
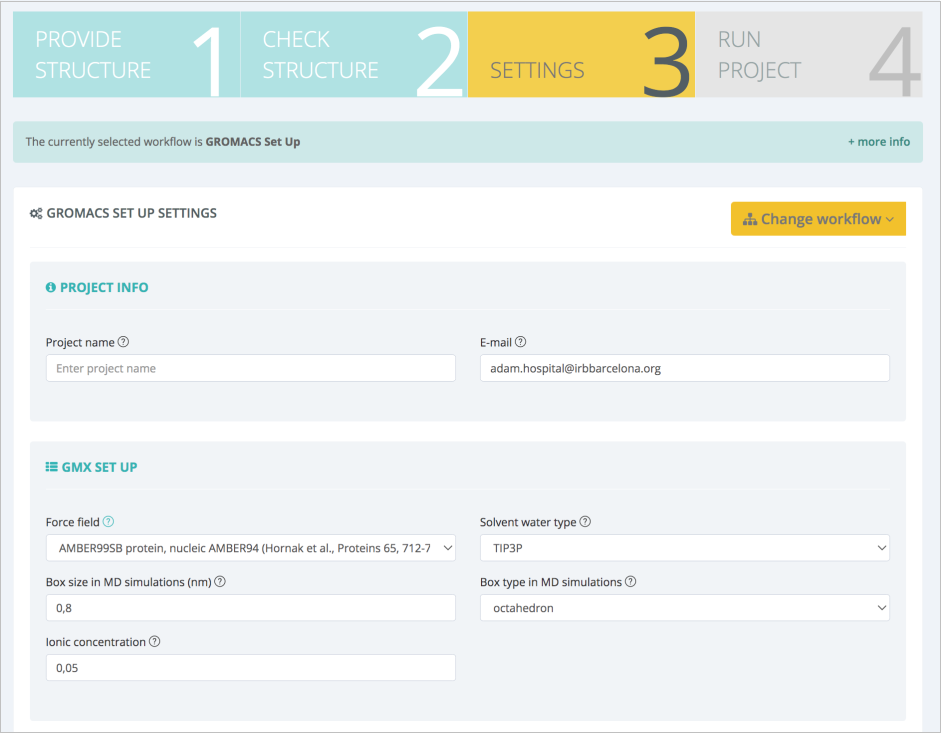
In this case, force field, water model, box type and size, and ionic concentration can be changed. Suggested default values are valid for our protein of interest: AMBER99SB force field (Lindorff-Larsen et al. Proteins, 2010), TIP3P water type (Price and Brooks. J Chem Phys, 2004), octahedral water box of an additional space of 8Å (0.8nm) added to the diameter of the system (largest distance between atoms) and an additional ionic concentration of 0.05 mol/l (after system neutralization). Next step summarizes the decisions made on the structure (ligands kept, structural issues solved/modified), and workflow settings chosen:
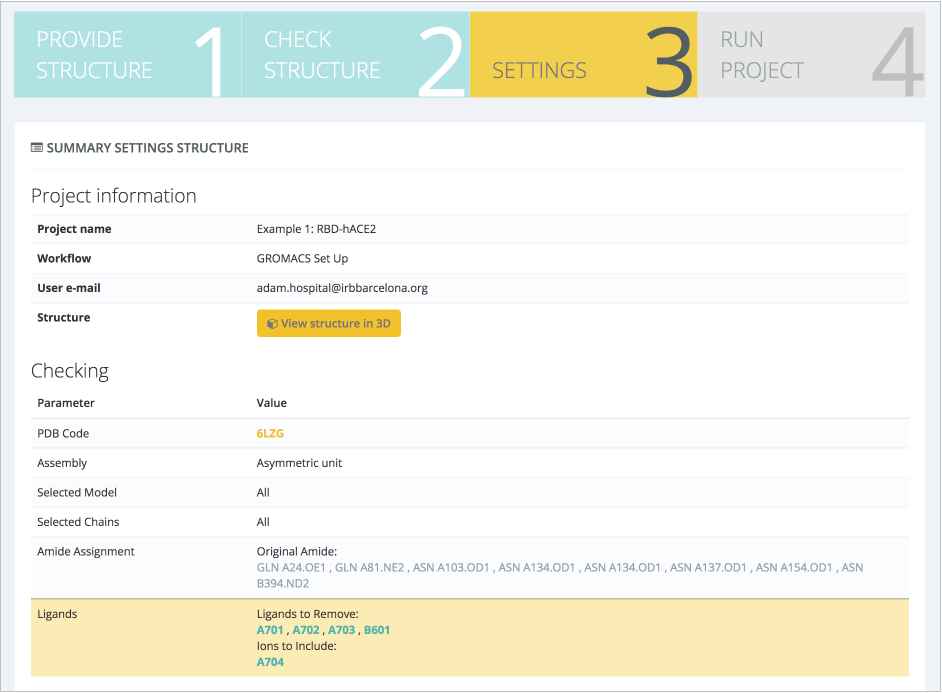
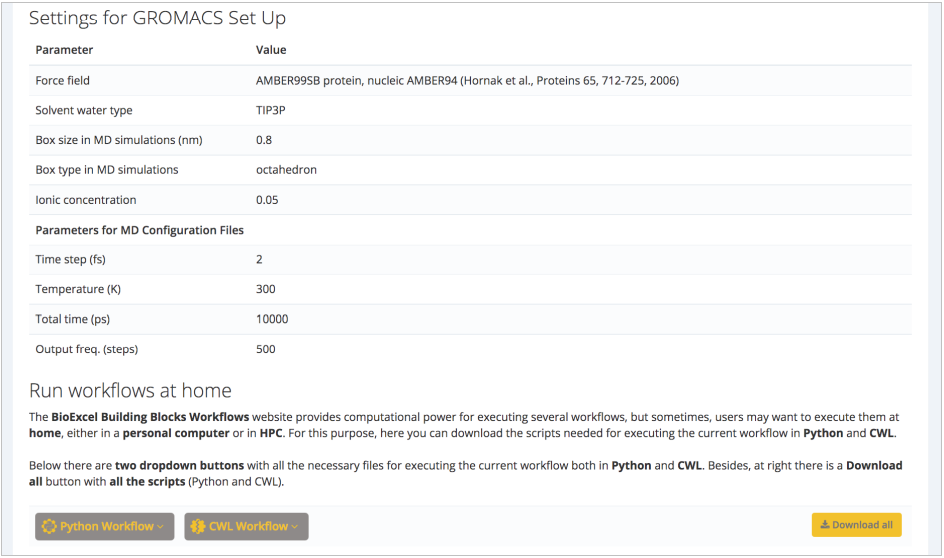
If the summary is not raising any possible mistake done in the previous steps, the pipeline execution can be launched. A progress of the workflow can be followed in real time:
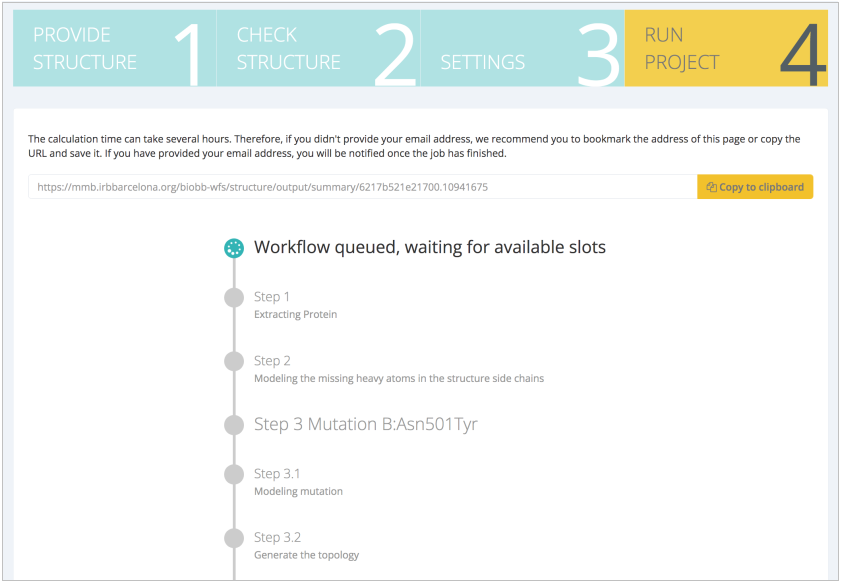
Otherwise, a URL link can be saved to get back to the execution later on. If the workflow has been launched from a registered user, it will always be available from the My Projects section.
The GROMACS Protein MD Setup with Mutations workflow runs a set of system preparation steps, including energy minimization, NVT and NPT equilibration processes. The progressive relaxation of the system can be visualized in the workflow output section through energy values along the different setup steps:
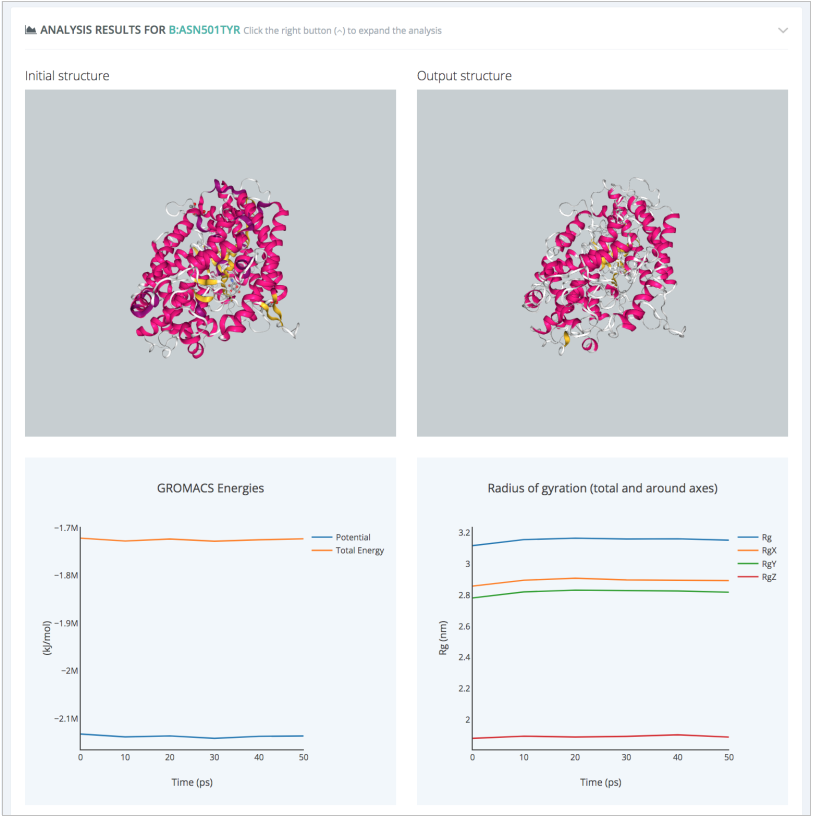
An additional system prepared to run MD simulations can be easily generated for the Wild Type protein complex using the GROMACS Protein MD Setup workflow, following the same steps, just excluding the residue mutation process. The final outcome of the study is a pair of MD simulations representing the dynamical behavior of the SARS-CoV-2 RBD-hACE2 complex with and without the N501Y mutation. Snapshots from the trajectories can be analysed to extract electrostatic and interaction pattern changes induced by the mutation.